ACIDES & BASES
Article modifié le
Théorie d'Arrhenius

Svante Arrhenius
Hulton Archive/ Getty Images

En 1887, un jeune chimiste suédois, Arrhenius, dans sa thèse de doctorat, proposa une théorie, alors révolutionnaire, pour expliquer les propriétés des solutions aqueuses d'électrolytes, en particulier leur conductibilité électrique : c'est la théorie de la dissociation ionique. Appliquée aux acides et bases, elle permit de préciser les notions précédentes et de les rendre quantitatives. Un acide HA est une substance qui, en solution aqueuse, fournit, lors de son équilibre de dissociation, des protons H+ :


À ces équilibres correspondent des constantes :
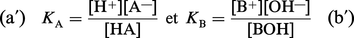
Pour chiffrer l'importance de la dissociation, on a introduit le degré de dissociation, α, qui est le rapport du nombre de molécules dissociées au nombre total de molécules ; si α est voisin de 1, on dit que l'acide ou la base est fort (HCl, KOH) ; s'il est faible, on dit que l'électrolyte est faible (acide acétique, ammoniaque NH4OH). Pour un acide ou une base de concentration globale C, il existe entre α et K la relation d'Ostwald :

La réaction de neutralisation d'un acide par une base (ou inversement) est facile à comprendre : l'addition des premiers membres de (a) et (b) représente l'action d'un acide sur une base. L'addition des seconds membres correspond à la formation de A— + B+, donc du sel AB fortement ionisé ; quant aux ions H+ et OH—, ils ne peuvent coexister en forte quantité, car ils se recombinent en eau : H2O est en effet un électrolyte très faible, et l'équilibre :


Les mesures
Les mesures quantitatives devinrent alors possibles à l'aide de la conductibilité puisque celle-ci croît avec α ; la comparaison, à concentration égale, des conductibilités d'acides ou bases en solution permit d'accéder à α, puis K, et d'établir ainsi une échelle de leurs forces. Un autre moyen d'évaluation de K, plus direct, fut fourni ensuite par les mesures de concentrations (ou activités) des ions H+, soit par potentiel d'électrodes, soit par indicateurs colorés. On peut y parvenir de deux façons différentes :
– Mesure de la concentration [H+]dans une solution d'acide à la concentration globale C ; il existe en effet entre α et [H+]la relation simple : [H+] = α C.
– Mesure de [H+]dans une solution d'acide HA en présence d'un de ses sels à la concentration [A—] ; la formule (a′) donne KA si [H+]est mesurable, puisque les concentrations [HA] et [A—]sont connues ; en particulier, si :
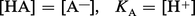
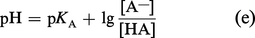
Les valeurs de pK varient beaucoup suivant les acides, les valeurs élevées correspondant aux acides les plus faibles : 10 pour les phénols, 9,2 pour l'acide borique, 4,6 pour l'acide acétique, 0,7 pour l'acide trichloracétique.
L'acide considéré peut renfermer plusieurs protons disponibles : par exemple H2SO4 (diacide), H3PO4 (triacide).
Pour un diacide H2A, il faudra considérer deux équilibres :

Avec H3PO4, par exemple, les pK1, pK2, pK3 successifs sont : 2,1 ; 7,2 et 12,3.
Cas des bases
Les raisonnements précédents sont applicables, en substituant les ions OH— aux ions H+. En solution aqueuse, on peut passer aisément de [H+]à [OH—], ce qui permet d'évaluer [OH—]. Considérons en effet l'équilibre :


À partir de (b′) on obtient :



Considérations résultant des notions précédentes
L'hydrolyse des sels
Dans l'eau pure, en vertu de l'équilibre (c), il existe autant d'ions H+ que d'ions OH—, et comme [H+][OH—] = 10—14, on en déduit [H+] = 10—7 = [OH—]soit pH = 7 ; au-dessous de ce pH, la solution est considérée comme acide, et au-dessus comme alcaline.
Une réaction telle que (d) résulte, comme il a été montré, de la superposition d'équilibres, elle devrait donc être elle-même équilibrée ; en réalité, elle est pratiquement complète dans le sens →, en raison de la faible dissociation de l'eau. L'action de l'acide chlorhydrique, acide fort, sur une égale quantité de soude, base forte, conduit bien en effet au chlorure de sodium et à l'eau, donc à une solution neutre, ce qui revient à dire que la réaction inverse, hydrolyse du sel AB (NaCl), est négligeable ; il arrive, cependant, qu'on doive la prendre en considération.
Supposons que HA soit un acide faible, et BOH une base forte ; la dissociation de l'eau ne sera pas négligeable, cette fois, par rapport à celle de HA. Donc, si l'on dissout le sel AB dans l'eau, on ne pourra éviter la coexistence de HA, acide faible, et OH—, dissociation de H2O, d'où l'équation (d) écrite en sens inverse :


L'hydrolyse de A— (sel AB du type acétate de sodium) donne des quantités égales de HA et OH—, mais, HA étant peu dissocié, la concentration de OH— prime celle de H+, et l'ensemble a un pH alcalin, qui peut devenir élevé : c'est le cas du carbonate de sodium qui donne, en raison du pK2 élevé de l'ion bicarbonate (10,2), un pH d'environ 12, ce qui faisait ranger primitivement les carbonates alcalins parmi les alcalis.
En effectuant les mêmes raisonnements sur un sel de base faible BOH et d'acide fort (NH4Cl), on assisterait à la réaction :

On y associe de même la constante d'hydrolyse :

Les indicateurs colorés
Il arrive que certains acides organiques faibles, dénommés HIn, soient fortement colorés, et que leurs formes ionisée In— et non ionisée HIn aient des couleurs très différentes, si bien que le rapport : [In—]/[HIn] pourra être déterminé par spectrophotométrie. La formule (e) montre que, si le pKA du colorant est connu, son introduction dans la solution, en très faible quantité pour ne pas altérer le pH, permettra d'évaluer celui-ci par spectrophotométrie, ou simple colorimétrie.
Une telle méthode, anciennement très employée, se trouva détrônée par la méthode potentiométrique, mais elle s'est révélée à nouveau très efficace pour chiffrer l'acidité des mélanges très acides (ou très alcalins), parce que la notion de pH y perd sa signification, et que de toute façon sa mesure y devient impossible. Pour de telles solutions on a introduit la fonction d'acidité H̄ (Hammett), définie de façon analogue au pH :

Cette grandeur mesure bien l'aptitude du milieu à échanger des protons ; par exemple, si son acidité croît, [HIn] augmente, [In—]diminue, et H̄ varie en conséquence ; dans les milieux ni trop acides ni trop alcalins, sa valeur est voisine de celle du pH.
Pour la mesurer, en milieu alcalin par exemple, dans les mélanges eau-éthylène diamine, on ajoute une petite quantité d'indicateur et on mesure la proportion de forme ionisée par spectrophotométrie dans un mélange « a » donné. Dans un mélange « b » plus riche en base, on mesure la proportion (plus forte) de forme ionisée, et de l'équation précédente, on tire :


L'équilibre In— — HIn s'observant en région très alcaline, on doit utiliser des indicateurs qui soient des acides très faibles. On opérerait de même dans les milieux fortement acides (mélanges eau-acide sulfurique) en utilisant des indicateurs qui soient des bases très faibles.
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Yves GAUTIER
: docteur en sciences de la Terre, concepteur de la collection
La Science au présent à la demande et sous la direction d'Encyclopædia Universalis, rédacteur en chef de 1997 à 2015 - Pierre SOUCHAY : professeur à l'université de Paris-VI-Pierre-et-Marie Curie et à l'École nationale supérieure de chimie, Paris
Classification
Médias

Acides et bases
Planeta Actimedia S.A.© Encyclopædia Universalis France pour la version française.
Svante Arrhenius
Hulton Archive/ Getty Images
Acides et bases
Encyclopædia Universalis France
Autres références
-
ACÉTIQUE ACIDE
- Écrit par Jacques METZGER
- 2 113 mots
- 3 médias
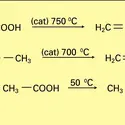
L'acide acétique, acide organique de formule CH3COOH, est le membre le plus important de la famille des acides carboxyliques. Il joue avec ses dérivés un rôle essentiel dans de nombreuses synthèses et dégradations biologiques accompagnant le métabolisme des aliments et la formation des tissus....
-
ACIDES-ALCOOLS
- Écrit par Jacques METZGER
- 1 300 mots
- 1 média

Un acide-alcool est une molécule renfermant au moins une fonction acide carboxylique et une fonction alcool.
La nature nous en fournit de nombreux exemples : acides lactique du lait aigri, malique des pommes avant maturité (diacide-monoalcool), tartrique du tartre des vins (diacide-diacool), citrique...
-
ALCOOLS
- Écrit par Jacques METZGER
- 5 834 mots
- 8 médias
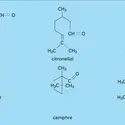
 Lafonction alcool présente à la fois un caractère acide et un caractère basique. Son acidité se manifeste lorsqu'il est mis au contact d'une base : le proton est cédé à cette dernière et l'anion alcoolate formé ainsi que l'acide conjugué de la base se trouvent en équilibre avec l'alcool non dissocié...
Lafonction alcool présente à la fois un caractère acide et un caractère basique. Son acidité se manifeste lorsqu'il est mis au contact d'une base : le proton est cédé à cette dernière et l'anion alcoolate formé ainsi que l'acide conjugué de la base se trouvent en équilibre avec l'alcool non dissocié... -
ALDÉHYDES ET CÉTONES
- Écrit par Jacques METZGER
- 7 356 mots
- 4 médias
 Les principales méthodes de préparation des dérivés carbonylés mettant en œuvre la réduction du substrat concernent les acides et les fonctions dérivées.
Les principales méthodes de préparation des dérivés carbonylés mettant en œuvre la réduction du substrat concernent les acides et les fonctions dérivées. - Afficher les 40 références
Voir aussi
- PHOSGÈNE (oxychlorure de carbone)
- ANHYDRIDE SULFUREUX ou DIOXYDE DE SOUFRE
- SELS
- NEUTRALISATION, chimie
- DISSOCIATION IONIQUE
- OSTWALD LOI DE DILUTION D'
- IONS
- HYDROLYSE
- INDICATEURS COLORÉS
- ÉLECTRONÉGATIVITÉ
- HYDROGÈNE LIAISON
- EAU-FORTE
- LEWIS THÉORIE DE
- ÉLECTROPHILES RÉACTIONS
- IZMAÏLOV RUSTUM GADJIEVITCH (1909- )
- COORDINENCE ou NOMBRE DE COORDINATION
- ALCALI
- pK
- RÉACTIONNELS MÉCANISMES
- BRØNSTED-LOWRY THÉORIE DE
- AMPHOTÈRE CORPS
- AMPHOLYTE
- VINAIGRE
- PYROSULFURIQUE ACIDE
- BJERRUM NIELS (1879-1958)
- BASE, chimie
- CONSTANTE DE DISSOCIATION
- IONISATION
- EAU, physico-chimie
- FORCE D'UN ACIDE ou D'UNE BASE
- ÉQUILIBRE, chimie
- CATALYSEURS
- HYDROXYLE ou OXHYDRYLE
- ADDITION, chimie
- POLARISATION, chimie
- CONCENTRATION, chimie
- AMIDURES
- ÉCHANGE CHIMIQUE
- CONSTANTE DIÉLECTRIQUE
- NUCLÉOPHILES SUBSTITUTIONS
- NATRON
- pH
- AUTOPROTOLYSE RÉACTION D'
- STÉRIQUE EFFET ou EMPÊCHEMENT
- CHIMIE HISTOIRE DE LA
- SOLUTION, chimie
- PLUIES ACIDES
- DÉPLACEMENT CHIMIQUE
- CONSTANTE D'ÉQUILIBRE, chimie
- ACIDITÉ ÉCHELLE D'
- DONNEUR, chimie
- ACCEPTEUR, chimie
- PEARSON RALPH GOTTFRIED (1919- )
