ACIDES & BASES
Article modifié le
Théorie de Brønsted-Lowry (1923)
Couples acidobasiques
Définition symétrique des acides et des bases
Elle définit comme acide toute substance capable d'émettre des protons H+. Ce sera le cas d'un acide faible, HA, comme dans l' équilibre (a), mais aussi de B+(NH4+), dans l'équilibre d'hydrolyse (g), la seule différence résidant dans la charge (0 ou + 1) de l'acide.
Inversement, une base sera toute substance capable de capter les protons : c'est non seulement le cas de BOH dans (g) lu dans le sens ←, mais aussi de l'ion A— (acétate) dans (a) lu dans le sens ←, la seule différence résidant dans la charge (0 ou — 1) de la base.

Il s'ensuit que dans un équilibre d'ionisation on ne peut concevoir l'existence de l'acide sans celle de la base correspondante : on dit que la paire HA—A— ou B+—BOH forme un couple acide-base. On notera qu'à un acide fort correspond une base faible (et inversement), car si HA est un acide fort, c'est qu'il perd facilement son proton, lequel inversement a peu tendance à réagir sur A—, qui se trouve donc être une base faible.
Relation entre constantes de dissociation acide et basique
Avec cette définition, la constante de l'acide sera :

Pour un acide du type HA, elle se confond avec l'ancienne définition (a′) ; avec un acide du type B+, elle n'est autre que l'ancienne constante d'hydrolyse (g′), B+ étant l'acide.
De même, si l'on adopte pour les bases la définition de l'ancienne théorie (capacité à émettre des ions OH—), non seulement les bases du type BOH rentrent dans cette catégorie (équations b et b′), mais aussi celles du type A—, en vertu de l'équilibre (f et f′), et la constante de la base sera :

Pour une base du type BOH, elle se confond avec l'ancienne définition ; avec une base de type A—, elle n'est autre que l'ancienne constante d'hydrolyse (f, f′). Il doit exister une relation entre KA et KB si l'acide et la base concernés appartiennent au même couple acidobasique ; en multipliant membre à membre les deux précédentes équations, on obtient KAKB = [H+][OH—] = Ke = 10—14.
Par exemple, la constante de dissociation de l'ammoniaque selon (b′) est 2 × 10—15 ; il revient au même de dire que NH4+ est un acide tel que :

Lorsqu'une espèce peut se comporter à la fois comme acide et base, on dit qu'elle est amphotère. Ainsi H2PO4— est capable de perdre un proton (acide) pour donner HPO42—, mais il peut en capter un (base) pour donner H3PO4, c'est un ampholyte.
On peut ainsi remarquer qu'il existe des acides à charges nulle (H3PO4), positive (NH+4) ou négative (H2PO4—).
Rôle du solvant
En raison de sa charge élevée par rapport à son volume, H+ n'existe pas comme tel en solution, mais se fixe sur le solvant (dénommé SH) qui se trouve jouer ainsi le rôle de base. Par conséquent, l'ionisation d'un acide dans l' eau ne s'écrira pas :




Parallèlement, l'ionisation de la base se fait suivant (f), l'eau se trouvant jouer le rôle d'acide, avec la constante de basicité :




Pour tout solvant SH autre que l'eau, on aurait pareillement :

Dans un solvant donné SH, un acide sera d'autant plus fort qu'il sera plus capable de céder des protons (constante élevée pour l'équilibre h), ce qui correspondra à une base d'autant plus faible, en vertu de KA KB = Ke.
Acidobasicité dans les solvants autres que l'eau
Les considérations précédentes montrent que la théorie de Brønsted, en faisant jouer un rôle symétrique aux acides et bases, permet de traiter de façon plus simple les échanges de protons, au sein des solvants capables d'échanger eux-mêmes des protons.
Dans ces conditions, pour uniformiser les notations, une base telle que NH4OH (NH3H2O) sera notée B et non BOH, et l'acide NH4+, dans NH4Cl, sera noté BH+ (NH3H+) et non B+ comme auparavant.
Les échanges de protons entre SH et le soluté acide ou basique dépendent de deux facteurs principaux.
Basicité (ou acidité) de SH
La constante de (h′) permet d'évaluer, dans l'eau, la force d'un acide ; elle est, d'ailleurs, la même que celle qui est donnée en (a′), la seule différence consistant à admettre que le proton est sous la forme H3O+ ; on aboutira ainsi à une classification des acides HA ou BH+ par force croissante.
Se reportant à (h), on voit que si HA est un acide suffisamment fort, toutes les molécules HA seront capables de fournir leurs protons à H2O, et HA sera un acide très fort (entièrement dissocié). Donc, au-delà d'une certaine force, il ne sera plus possible de différencier les acides ; c'est ainsi que, malgré leurs forces décroissantes dans l'ordre HClO4 > HI > HBr > HNO3 > HCl, ces acides, entièrement dissociés dans l'eau, y sont également forts : on dit que l'eau exerce un « effet nivelant ». Il n'existe d'ailleurs pas, dans ce solvant, d'acide plus fort que H3O+, car il réagirait sur H2O pour donner ce dernier ion.
En revanche, dans l'alcool, solvant moins basique que l'eau, les acides chlorhydrique et nitrique ne sont plus entièrement dissociés et peuvent être différenciés des acides plus forts. Au contraire, dans l'ammoniac liquide, solvant beaucoup plus basique que l'eau, un acide aussi faible que l'acide acétique est entièrement dissocié.
Des considérations analogues interviennent dans le domaine basique : dans l'eau, des bases très fortes telles que les ions éthylate (C2H5O—) et amidure (NH2—) déplacent entièrement les équilibres tels que :

On voit qu'en raison de leur effet nivelant plus ou moins prononcé, les solvants exercent une profonde influence sur l'utilisation des réactifs : l'exemple précédent montre qu'il est inutile d'effectuer des réactions avec NH2— dans l'eau.
Si nous désirons un solvant pouvant mettre en évidence des différences dans les tendances des divers acides à donner des protons, il devra lui-même avoir peu tendance à capter des protons (c'est-à-dire que SH2+ sera un acide très fort).
Ainsi, l'ammoniac est un solvant nivelant pour les acides, car tous sont ramenés à la force de l'acide faible NH4+ ; par contre, NH2— étant une base très forte (car les propriétés acides de NH3 sont faibles), l'ammoniac permettra de différencier les bases, même fortes.
Constante diélectrique ε de SH
Les forces de Coulomb s'exerçant entre deux charges Z1 et Z2, distantes de x, sont de la forme : Z1Z2/εx2. Il s'ensuit que le travail nécessaire pour séparer les charges, dans une ionisation du type (i), sera d'autant plus faible (donc l'ionisation de HA plus facile) que ε augmentera : ainsi, à basicité égale du solvant, HA sera plus fort dans un solvant à ε élevée. À partir de l'expression précédente du travail, on peut montrer que pKHA est de la forme Cte = a/ε, où a est de l'ordre de 150.
Si l'acide est du type BH+, la réaction d'ionisation :


Précisons que toutes les considérations précédentes sont valables pour des constantes diélectriques suffisantes (ε > 20) et seulement de façon approximative, car nous n'avons envisagé que l'action des deux principaux facteurs. Aux faibles valeurs de ε, l'énergie nécessaire à la séparation des ions est trop élevée, et ces ions restent unis, par attraction électrostatique, sous forme de paires d'ions. Une paire H+A— est donc différente d'une molécule HA, où la liaison est covalente, mais les méthodes expérimentales permettent difficilement d'opter entre les deux possibilités.
Comparaison des acidités (ou basicités) dans divers solvants
Influence de la basicité
Considérons deux acides du type BH+, s'ionisant dans un solvant SH suivant :

Il est facile de constater, par soustraction membre à membre, que ces deux équilibres ne sont pas indépendants de :

Comme cet équilibre ne fait pas figurer SH, il doit en être indépendant, ainsi que K1/K2, et pK1 — pK2 : donc, si seule la basicité de SH est en cause, les différences de pK entre divers couples acide-base BH+ — B restent les mêmes dans tous les solvants.
Il s'ensuit que les différences de pK des acides BH+ dans deux solvants (l'un d'eux étant souvent l'eau, prise comme solvant de référence) doivent être les mêmes pour tous : le pK d'un acide dans SH se déduit du pK dans l'eau par une constante additive ΔpK+ (positive si SH est plus acide que H2O).
Influence de ε
Ces considérations sont valables pour les acides du type HA : le pK d'un acide HA dans SH se déduit du pK dans l'eau par une constante additive ΔpK0.
Néanmoins, comme pKAH varie avec ε, et que pKBH+ varie peu, il s'ensuit que ΔpK+ et ΔpK0 seront différents. Donc les écarts des pK entre deux acides du type BH+ ou du type HA ne varient pas avec SH, mais les écarts entre deux acides des types HA et BH+ varient avec SH.
Échelle d'acidité
Il sera possible de classer, dans l'eau, les acides par pK croissant établissant ainsi une échelle d'acidité ; en raison de l'effet nivelant, cette échelle sera limitée des deux côtés. Compte tenu de ce qu'on ne rencontre que peu fréquemment des acidités ou alcalinités fortes, supérieures à 1 mole par litre, ce qui correspond à des pH extrêmes de 0 à 14, l'échelle d'acidité en milieu aqueux s'étendra de pK = 0 à pK = 14.
Dans un autre solvant SH, il sera possible, en utilisant une échelle de pSH2+ analogue au pH (on devrait dire pH3O), de classer également les acides par pK croissants, pourvu que les pK définis dans l'échelle pSH2+ soient compris entre 0 et — log Ke (Ke, produit ionique de SH, 10—14 dans l'eau).
Pour passer de l'échelle dans H2O à l'échelle dans SH, il suffit d'une translation ΔpK+ pour les acides BH+, et ΔpK0 pour les acides HA.
Inversement, connaissant pKSH, on passera à pKH2O par une même translation de signe contraire. Il suffit donc de tracer une échelle unique dans l'eau, pour en déduire celles qui sont relatives aux autres solvants moyennant la connaissance, pour chaque solvant, de ΔpK0 et ΔpK+.
Comme prévu, les acides de pKH2O inférieur à 0 ou supérieur à 14 figurant sur cette échelle ne pourront se différencier dans l'eau, mais les valeurs données serviront, en utilisant l'opération que nous venons d'indiquer, à établir le pK dans un solvant SH.

Échelle d'acidité
Encyclopædia Universalis France
La figure 1 donne l'exemple de telles échelles. Deviennent différenciables dans le méthanol tous les acides qui, dans l'échelle de l'eau, possèdent un pK supérieur à — 1,3 pour BH+, et à — 4,9 pour HA ; on constate alors que les acides iodique et surtout nitrique ne sont plus des acides forts.
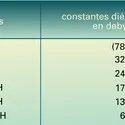
De plus, Ke = 10—16,7, l'échelle d'acidité est comprise entre pSH2+ = 0 et 16,7, ce qui correspond, dans l'eau, à des pK compris entre — 4,9 et 11,8 : le méthanol ne peut différencier les acides de pK supérieurs à 11,8 dans l'eau.
De telles échelles permettent de prévoir les réactions acides-bases ; une réaction telle que (j) :

Ainsi la figure 1b montre que l'acide fluorhydrique réagira quantitativement sur l'ammoniac :

Lorsque le pK est extérieur aux limites de l'échelle, il y aura réaction totale de l'un des partenaires avec SH :

Réactions 7 à 14
Encyclopædia Universalis France
– si le pK dépasse la limite supérieure, ce sera le partenaire basique (réaction 7) ;
– s'il est au-dessous de la limite inférieure, ce sera le partenaire acide (réaction 8).
Le méthanol diffère peu de l'eau au point de vue acidobasique. Il n'en est pas de même pour l'ammoniac liquide et l'acide sulfurique, solvants respectivement plus basique et plus acide que l'eau.
Solvant plus basique que l'eau : l'ammoniac liquide
La structure de l'ammoniac liquide, qui bout à — 33 0C, rappelle beaucoup celle de l'eau, en raison de l'existence de liaisons hydrogène. Il s'ionise suivant :

Tous les acides plus forts que l'ion ammonium NH4+ sont ici des acides forts (cf. H3O+ dans l'eau) ; les amidures NH2— sont les analogues des hydroxydes dans l'eau. La réaction typique de neutralisation, est, ici,

Les métaux sont de même attaqués par les sels d'ammonium avec dégagement d'hydrogène :

Dans les réactions où une base très forte est nécessaire, on utilisera surtout l'amidure de potassium (ceux des autres métaux sont très peu solubles dans l'ammoniac liquide).
On obtient, par action des sels métalliques, des réactions rappelant l'obtention des hydroxydes et oxydes dans l'eau :

L'amidure, base très forte, permet l'obtention de sels d'acides très faibles :


Solvant plus acide que l'eau : l'acide sulfurique
Les associations entre molécules de solvant sont ici très fortes, comme en témoignent les viscosité et température d'ébullition élevées.
Pour passer de l'échelle de l'eau (pH3O+) à celle de l'acide sulfurique (pSO4H3+), il faut ajouter 12,5 pour les acides BH+ et 13,5 pour les acides HA. La conductivité, relativement élevée, indique un produit ionique fort :


On ne pourra différencier que les acides donnant un pSO4H3+ compris entre 0 et 3,6, ce qui correspond, en adoptant l'échelle dans l'eau aux limites — 12,5 (ou — 13,5) à — 8,9 (ou — 9,9) ; c'est-à-dire que de très nombreuses substances se comportent comme des bases ; ainsi l'eau y acquiert des propriétés nettement basiques :


En particulier, l'ion Cl— est transformé en acide chlorhydrique non dissocié :


Outre l'équilibre de dissociation ionique (k), apparaît également dans l'acide sulfurique lui-même l'équilibre :

L'action de l'eau a été bien mise en évidence par cryoscopie : les premières additions ne font varier qu'assez peu le point de fusion (primitivement de 10,5 0C) ; elles font apparaître en effet des ions HSO4—, qui rétrogradent l'auto-ionisation par déplacement de l'équilibre (k).
Quand on a ajouté la faible quantité nécessaire pour que le déplacement soit total, le point de fusion décroît régulièrement, mais son abaissement est le double de celui qu'on pourrait prévoir s'il s'agissait d'une simple dissolution. Cela n'est pas étonnant, puisque l'abaissement est proportionnel à la concentration en particules dissoutes, et que l'introduction d'une particule H2O provoque la formation de deux particules, H3SO4+ et HSO4—.
Catalyse acido-basique
Certaines réactions sont accélérées par la présence de substances étrangères (fréquemment il suffit de très faibles quantités), que l'on trouve inchangées en fin de réaction et que l'on nomme catalyseurs. Lorsqu'en solution le catalyseur est constitué par les ions H+ ou OH—, on dit qu'il y a « catalyse acidobasique ». Un des exemples les plus anciennement connus est l'inversion du sucre (transformation en glucose et fructose) catalysée par les acides forts.
La vitesse d'une telle réaction est normalement proportionnelle à la concentration de substance restant en solution, et la constante k de proportionnalité s'appelle constante de vitesse. Dans l'exemple ci-dessus, k reste effectivement constant pendant que s'effectue la réaction, mais dépend de l'acidité au départ, qui d'ailleurs ne varie pas dans le cours de la réaction (contrairement à la concentration du sucre, qui décroît constamment, puis finit par s'annuler). On écrira ainsi : k = kH[H+].
D'autres réactions sont catalysées par les ions OH— ; il arrive qu'une même réaction soit catalysée par ces deux ions, selon des mécanismes d'ailleurs différents ; on a alors

Puisque [H+]et [OH]sont liées par la relation Ke = [H+][OH—], il est plus logique d'exprimer k en fonction de [H+]seulement :

Les deux derniers termes du second membre varient en sens inverse en fonction du pH ; k passera donc par un minimum pour un certain pHm obtenu en annulant la dérivée de k :

Mécanisme
Puisqu'on retrouve le catalyseur inchangé en fin de réaction, et qu'il accélère néanmoins celle-ci, il faut bien admettre qu'à une certaine étape il cède (ou enlève) des protons à la substance réagissante, qui doit donc posséder des propriétés faiblement basiques (ou acides), même si elles sont si faibles que les méthodes classiques ne peuvent les déceler.
Le composé intermédiaire formé, de structure différente de la substance de départ, peut se montrer facilement attaquable par les réactifs, en régénérant, entre autres, les ions H+ (ou OH—) ; même s'il n'existe qu'en faible concentration, il se reforme continuellement à mesure qu'il réagit, et la réaction finale progressera très rapidement.
Ainsi, dans la catalyse acide de l'hydrolyse des esters R′—COOR, le proton donne naissance à un carbocation R′—C+(OH)OR très sensible à une attaque par l'eau, qui rompt la liaison O—R en donnant l'acide R′COOH, l'alcool ROH et le proton de départ.
De même, les bases accélèrent les réactions de nombreuses méhylcétones, en captant un proton et formant un ion énolate à réactivité élevée, grâce à la double liaison entre les carbones.
Catalyse généralisée
On supposa primitivement que les ions H+ ou OH— étaient les seuls catalyseurs effectifs lors de la catalyse par les acides ou les bases. Tant que l'ion hydrogène fut regardé comme un proton libre H+, il semblait légitime de lui attribuer un pouvoir catalytique particulier, ne fût-ce qu'en raison de son champ électrostatique intense, mais on sait maintenant qu'il est sous forme H3O+ dans l'eau. Dans ces conditions, il ne se différencie d'aucun autre acide de Brønsted, et toute particule autre que H3O+, susceptible de libérer des protons, peut se montrer catalytiquement active, quelle que soit sa charge (CH3CO2H ; NH4+, H2PO4—).
Du côté alcalin, ces considérations subsistent : l'ion OH—, qui n'est autre que l'anion d'un acide faible (H2O), ne doit pas obligatoirement jouer un rôle privilégié par rapport à d'autres bases ou anions accepteurs de protons (NH3, CH3CO2—, PO43—). Ainsi, de nombreuses réactions, catalysées par les acides ou les bases relèvent de la catalyse généralisée, dans laquelle les espèces catalysantes sont non seulement H3O+ ou OH—, mais tout acide ou base selon Brønsted. Par conséquent, dans un milieu renfermant un acide faible AH, et son sel A— (base), on devra écrire la formule 9 plus générale.
Assez souvent, quelles que soient les conditions, le terme en kHA ou kA— est continuellement négligeable devant celui en kH ou kOH. Cependant, même s'il n'en est pas ainsi, il peut arriver que seules H3O+ et OH— interviennent dans l'expression de la vitesse, quoique les autres espèces puissent être actives. Dans ces deux cas, on dit qu'il y a catalyse spécifique par les ions H3O+ (ou OH—). Pour savoir s'il y a catalyse généralisée ou non, le mieux est d'effectuer la réaction dans des mélanges HA + A— dont le rapport [HA]/[A—]est maintenu constant, ce qui entraîne la constance de [H3O+]. En faisant varier, à rapport constant la concentration des constituants, la vitesse de réaction doit rester la même si la catalyse est spécifique, sinon on observe une variation dont la grandeur permet d'évaluer kA— et kHA.
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Yves GAUTIER
: docteur en sciences de la Terre, concepteur de la collection
La Science au présent à la demande et sous la direction d'Encyclopædia Universalis, rédacteur en chef de 1997 à 2015 - Pierre SOUCHAY : professeur à l'université de Paris-VI-Pierre-et-Marie Curie et à l'École nationale supérieure de chimie, Paris
Classification
Médias

Acides et bases
Planeta Actimedia S.A.© Encyclopædia Universalis France pour la version française.

Svante Arrhenius
Hulton Archive/ Getty Images
Acides et bases
Encyclopædia Universalis France
Autres références
-
ACÉTIQUE ACIDE
- Écrit par Jacques METZGER
- 2 113 mots
- 3 médias
L'acide acétique, acide organique de formule CH3COOH, est le membre le plus important de la famille des acides carboxyliques. Il joue avec ses dérivés un rôle essentiel dans de nombreuses synthèses et dégradations biologiques accompagnant le métabolisme des aliments et la formation des tissus....
-
ACIDES-ALCOOLS
- Écrit par Jacques METZGER
- 1 300 mots
- 1 média

Un acide-alcool est une molécule renfermant au moins une fonction acide carboxylique et une fonction alcool.
La nature nous en fournit de nombreux exemples : acides lactique du lait aigri, malique des pommes avant maturité (diacide-monoalcool), tartrique du tartre des vins (diacide-diacool), citrique...
-
ALCOOLS
- Écrit par Jacques METZGER
- 5 834 mots
- 8 médias
 Lafonction alcool présente à la fois un caractère acide et un caractère basique. Son acidité se manifeste lorsqu'il est mis au contact d'une base : le proton est cédé à cette dernière et l'anion alcoolate formé ainsi que l'acide conjugué de la base se trouvent en équilibre avec l'alcool non dissocié...
Lafonction alcool présente à la fois un caractère acide et un caractère basique. Son acidité se manifeste lorsqu'il est mis au contact d'une base : le proton est cédé à cette dernière et l'anion alcoolate formé ainsi que l'acide conjugué de la base se trouvent en équilibre avec l'alcool non dissocié... -
ALDÉHYDES ET CÉTONES
- Écrit par Jacques METZGER
- 7 356 mots
- 4 médias
 Les principales méthodes de préparation des dérivés carbonylés mettant en œuvre la réduction du substrat concernent les acides et les fonctions dérivées.
Les principales méthodes de préparation des dérivés carbonylés mettant en œuvre la réduction du substrat concernent les acides et les fonctions dérivées. - Afficher les 40 références
Voir aussi
- PHOSGÈNE (oxychlorure de carbone)
- ANHYDRIDE SULFUREUX ou DIOXYDE DE SOUFRE
- SELS
- NEUTRALISATION, chimie
- DISSOCIATION IONIQUE
- OSTWALD LOI DE DILUTION D'
- IONS
- HYDROLYSE
- INDICATEURS COLORÉS
- ÉLECTRONÉGATIVITÉ
- HYDROGÈNE LIAISON
- EAU-FORTE
- LEWIS THÉORIE DE
- ÉLECTROPHILES RÉACTIONS
- IZMAÏLOV RUSTUM GADJIEVITCH (1909- )
- COORDINENCE ou NOMBRE DE COORDINATION
- ALCALI
- pK
- RÉACTIONNELS MÉCANISMES
- BRØNSTED-LOWRY THÉORIE DE
- AMPHOTÈRE CORPS
- AMPHOLYTE
- VINAIGRE
- PYROSULFURIQUE ACIDE
- BJERRUM NIELS (1879-1958)
- BASE, chimie
- CONSTANTE DE DISSOCIATION
- IONISATION
- EAU, physico-chimie
- FORCE D'UN ACIDE ou D'UNE BASE
- ÉQUILIBRE, chimie
- CATALYSEURS
- HYDROXYLE ou OXHYDRYLE
- ADDITION, chimie
- POLARISATION, chimie
- CONCENTRATION, chimie
- AMIDURES
- ÉCHANGE CHIMIQUE
- CONSTANTE DIÉLECTRIQUE
- NUCLÉOPHILES SUBSTITUTIONS
- NATRON
- pH
- AUTOPROTOLYSE RÉACTION D'
- STÉRIQUE EFFET ou EMPÊCHEMENT
- CHIMIE HISTOIRE DE LA
- SOLUTION, chimie
- PLUIES ACIDES
- DÉPLACEMENT CHIMIQUE
- CONSTANTE D'ÉQUILIBRE, chimie
- ACIDITÉ ÉCHELLE D'
- DONNEUR, chimie
- ACCEPTEUR, chimie
- PEARSON RALPH GOTTFRIED (1919- )
