CATALYSE
Article modifié le
Particularités des catalyseurs solides
La catalyse étant un phénomène de surface, l'activité d'un solide sera en général d'autant plus grande que le rapport surface/masse, ou aire spécifique (m2/g) sera plus élevé, c'est-à-dire que le solide sera plus poreux et divisé. L'art de la préparation des catalyseurs contient de nombreuses recettes à cette fin. En général, le solide sera d'autant plus divisé qu'il sera élaboré à une température plus basse : on pourra opérer par voie humide en formant un précipité à partir d'une solution, ou par voie sèche en attaquant un solide par un gaz. Les moyens mécaniques de broyage sont assez peu utilisés. À titre d'exemple, l'aire spécifique d'un catalyseur peut aller de quelques m2/g à plusieurs centaines de m2/g :
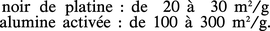
Pour les solides divisés non poreux, l'aire spécifique est en relation simple avec la dimension moyenne des grains. Ainsi, pour le platine de masse volumique 21,45 g/cm3, en supposant les grains cubiques, l'arête moyenne des cubes serait de 10,0 à 14,5 nm dans l'exemple cité, soit 35 à 50 atomes. On peut diminuer fortement ces dimensions en dispersant le métal sur un support poreux comme une alumine, et obtenir ainsi des aires spécifiques de 50 à 100 m2/g de platine, soit des tailles de 2,5 à 5,5 nm. Pour les solides poreux, l'aire spécifique, qui peut atteindre plusieurs milliers de m2/g, est liée à la finesse des pores, qui peuvent descendre à quelques nanomètres. Une grande aire spécifique est d'autant moins nécessaire que le catalyseur est plus actif, et il est possible, par exemple, dans certaines réactions, d'utiliser le platine en fils (toiles).

Mais l'aire spécifique donne la quantité de surface active et non sa qualité, c'est-à-dire la nature et la densité des centres actifs. Cette qualité dépend énormément de la méthode d'élaboration du catalyseur. Par définition, les centres actifs sont les atomes ou groupes d'atomes de la surface les plus propres à former des combinaisons chimiques, et, en l'absence de précautions spéciales, ils se trouveront tous saturés lors de la préparation du catalyseur.
En effet, le solide trouvera toujours, soit dans la solution mère, soit dans l'atmosphère qui l'entoure, des molécules disposées à se combiner avec les atomes de sa surface. Par exemple, un solide tel que le nickel divisé adsorbera avec beaucoup de ténacité l'oxygène de l'air, et les centres acides d'une alumine préparée par voie aqueuse seront neutralisés par les ions basiques.
Le traitement préliminaire activant qui est, par exemple, une réduction par l'hydrogène, ou une déshydratation par chauffage, sera donc essentiel.
Il est clair que la simple spécification de la nature chimique d'un catalyseur est très insuffisante pour le caractériser, car ses propriétés finales dépendront, comme on l'a vu, de tous les processus employés pour sa fabrication.
La durée de vie d'un catalyseur solide est limitée par celle de ses centres actifs. Leur disparition (désactivation) peut avoir de nombreuses causes. Parmi les plus fréquentes, on citera la chute de l'aire spécifique par coalescence des grains, ou frittage, provoqué par une température trop élevée : c'est un processus physique irréversible, qui est dû à l'instabilité thermodynamique des petits grains par rapport aux gros (énergie superficielle).
L'empoisonnement du catalyseur se produit lorsqu'on introduit accidentellement dans le milieu un corps, dit poison, dont les molécules sont très fortement adsorbées par les centres actifs, qui se trouvent ainsi bloqués. Les composés du soufre (hydrogène sulfuré, sulfure de carbone, thiols) sont ainsi des poisons violents pour les métaux (Ni, Pt).
L'encrassement du catalyseur est observé lorsqu'une réaction secondaire forme à sa surface des produits indésorbables de grande masse moléculaire. Ainsi, dans de nombreux traitements catalytiques des hydrocarbures, des produits lourds, mal définis sous le nom de coke, bloquent lentement les centres actifs. L'empoisonnement et l'encrassement sont dits réversibles lorsque par un traitement approprié (autre que la destruction totale du catalyseur en ses éléments) on peut débloquer les centres actifs mis hors d'usage. Ainsi, un dépôt de coke peut être enlevé par combustion ménagée à l'oxygène dilué, suivi éventuellement d'une réduction, si on a un métal dans le catalyseur.
Composition et aspect des catalyseurs solides
Les catalyseurs utilisés en pratique sont fréquemment des mélanges où l'on peut distinguer le composant actif, le support et les promoteurs.
Ces derniers sont des composants par eux-mêmes inactifs, mais capables, par divers moyens, d'améliorer la stabilité, la quantité ou la qualité des centres actifs.
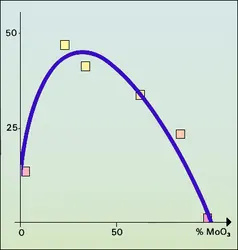
Effet promoteur
Encyclopædia Universalis France
Certains empêchent le frittage de la phase active et conservent une aire spécifique élevée par simple insertion entre les grains de cette phase : ainsi, un faible pourcentage d'oxyde de chrome ou d'aluminium prolonge considérablement la vie du nickel divisé. D'autres se combinent ou se dissolvent dans la phase active et modifient la nature même des centres actifs, par un effet qui n'est pas sans analogie avec celui du changement des ligandes inertes dans les catalyseurs solubles (cf. Catalyseurs complexes). Il y a promotion lorsque cette addition augmente l'activité ou la sélectivité du catalyseur, et l'on observe toujours un maximum de ces grandeurs quand la dose de promoteur augmente (fig. 5). Lorsque la quantité optimale de promoteur est élevée, on parle de catalyseur mixte : ainsi, les oxydes mixtes de vanadium et de molybdène ont une sélectivité maximale pour l'oxydation du benzène en anhydride maléique (base de matières plastiques du type polyester) pour environ V2O5/MoO3 = 3.
Les catalyseurs se présentent en poudres, grains, sphérules, ou pastilles de dimensions variant de quelques centaines de microns à plusieurs centimètres. Les plus gros sont employés en lit fixe, couches d'épaisseur variable, traversées par le fluide, liquide ou gaz, tandis que les plus fins sont mis en suspension dans ce fluide, dont il faut les séparer (décantation, filtration...). Dans tous les cas, les produits de l'opération sont plus faciles à séparer du catalyseur que dans la catalyse homogène.
En pratique, la durée de vie du catalyseur est très importante, car elle règle l'incidence du prix du catalyseur sur les prix de revient de la fabrication. Il résulte de ce qui précède que cette durée de vie augmentera si on abaisse la température de travail et si on augmente la pureté des corps introduits.
La consommation de catalyseur par tonne de produit traité (pertes ou désactivation irréversible) peut varier de 1 kg (craquage catalytique) à 1 g environ (synthèse de l'ammoniac).
Cinétique de la catalyse hétérogène
La cinétique des réactions catalysées est dominée par la vitesse de réaction des centres actifs avec les corps de départ ou la vitesse de libération de ces centres par désorption des produits ; ces deux vitesses étant égales lorsque s'est établi le régime stationnaire, l'étape la plus lente gouverne la vitesse globale. Cette compétition pour les centres actifs se traduit souvent par un effet de ralentissement de la réaction par ses propres produits, appelé auto-inhibition.
Ainsi, la synthèse de l'ammoniac est inhibée par l'ammoniac, et sa décomposition est inhibée par l'hydrogène :

Notons que, conformément à la thermodynamique (cf. supra), on a la condition d'équilibre v = 0 :

La phase fluide n'intervient que comme un réservoir de réactifs et de produits, qui sont respectivement amenés vers la surface, ou en sens inverse, par la diffusion des molécules. Si la réaction de surface est très rapide, la vitesse globale sera limitée par celle de ces transferts et, à la limite, deviendra indépendante de l'activité du catalyseur. Cette limitation diffusionnelle joue un grand rôle lorsque les centres actifs sont sur les parois internes de pores très fins (micropores de quelques dizaines d'angströms de diamètre), à travers lesquels la diffusion est lente. La partie centrale d'un grain de catalyseur microporeux risque ainsi de ne pas être du tout utilisée, les molécules des réactifs étant complètement transformées avant de les atteindre. Une aire spécifique trop élevée n'est pas toujours avantageuse, notamment pour les catalyseurs très actifs. L'activité sera mesurée ici en moles converties par unité de temps et unité de surface active. Quand la chose est possible, la densité superficielle de centres actifs est une indication très précieuse. Elle est d'environ 1015 par cm2 sur les métaux (Ni, Pt, Fe) et de 1012 par cm2 sur les oxydes métalliques. On peut l'estimer par des mesures d'adsorption chimique.
Les grandes familles de catalyseurs solides
Les catalyseurs solides sont en général des métaux ou des composés binaires ou ternaires de ces métaux avec des éléments non métalliques et, de façon plus générale, des solides à point de fusion élevé, indiquant de fortes liaisons chimiques entre leurs atomes ou ions. Comme le note Langmuir (cf. chap. 4, Catalyse hétérogène), le clivage de tels solides crée une surface hautement insaturée au point de vue chimique, et, partant, des centres actifs. On trouve assez souvent que ces centres actifs ont des propriétés semblables à celles de certains catalyseurs homogènes, et accélèrent les mêmes réactions.

Structures octaédriques
Encyclopædia Universalis France
Une première classe est constituée par les solides qui activent l'hydrogène, l'oxyde de carbone, l'éthylène, l'oxygène : ce sont les métaux de transition et leurs oxydes, sulfures... qui donnent ainsi les complexes solubles ayant des propriétés catalytiques similaires. Leurs centres actifs sont construits autour de l'atome de transition, chargé ou non, qui permet la formation d'un grand nombre de liaisons (coordinance variable). Que cet atome soit au centre d'une molécule complexe ou dans un solide, sa configuration électronique reste la même ; aussi, une molécule comme CO liée dans un métal-carbonyle ou à un atome de surface du métal cristallisé, ou à un ion superficiel de son oxyde se trouve-t-elle dans un état actif (fig. 6). Les différences constatées peuvent s'expliquer par l'influence des ligandes inertes ou des atomes qui les remplacent dans les solides.
On trouve dans cette classe les catalyseurs d' hydrogénation, comme le nickel, le platine, le fer, le cobalt, l'oxyde de nickel, l'oxyde de chrome, etc., qui sont aussi des catalyseurs de déshydrogénation et d'hydrogénolyse, car ils activent notamment les liaisons H-H, C-H, O-H, N-H en séparant le H.
Les catalyseurs d'oxydation, qui activent le dioxygène O2 tels que le platine, les oxydes de manganèse, cobalt, cuivre, vanadium, molybdène, tungstène appartiennent également à cette classe. Leurs centres actifs se combinent aussi volontiers avec les corps organiques insaturés : éthylène, benzène et dérivés, et permettent leur hydrogénation et leur oxydation.

Structures tétraédriques
Encyclopædia Universalis France
Une deuxième classe comprend les oxydes acides ou basiques dont l'activité catalytique est semblable à celle des acides ou bases solubles, et qui activent les molécules ionisables, par exemple par addition ou soustraction d'un proton H+. On peut d'ailleurs titrer l'acidité des catalyseurs solides par les mêmes indicateurs colorés d'emploi courant en solution aqueuse, et mesurer leur force. Les centres acides à la surface des catalyseurs mixtes silice-alumine sont aussi forts que l'acide sulfurique à 90 p. 100 : cela s'explique par la présence de groupes fonctionnels semblables aux anions des oxyacides solubles. L'analogie entre AlO4— (sur les silices-alumines) et ClO4— (solutions d'acide perchlorique) mérite d'être soulignée (fig. 7).
Quant aux centres basiques, ils peuvent être identifiés aux anions de surface, par exemple O2—, dans les oxydes, lorsque ces anions sont incomplètement saturés.
Cette classe inclut un grand nombre de silicates, phosphates, borates, les alumines, la thorine. Le groupe des silicates d'aluminium synthétiques ou naturels est particulièrement riche ; leurs centres actifs acides sont les groupes AlO4— H+ dus à la dispersion de l'aluminium dans un réseau de silice. Les argiles naturelles activées, les tamis moléculaires naturels ou artificiels en font partie.
L'activation de l'eau et des oléfines permet de réaliser les réactions d'hydratation, de déshydratation, d'alcoylation, de craquage, de polymérisation, d'isomérisation des corps organiques.
En conclusion, il faut souligner l'importance pratique de la catalyse dans l'industrie chimique et donc dans la vie économique moderne. La découverte d'un catalyseur nouveau peut bouleverser la structure de toute une industrie, voire même le cours de notre civilisation. À ce titre, le catalyseur est vraiment la version moderne de la pierre philosophale.
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Henri Jean-Marie DOU : ingénieur I.P.S.O.I., docteur ès sciences, directeur de recherche au C.N.R.S., au Centre de recherche rétrospective de Marseille, président de la Société française de bibliométrie appliquée
- Jean-Eugène GERMAIN : professeur de chimie à l'université de Lyon-I-Claude-Bernard
Classification
Médias
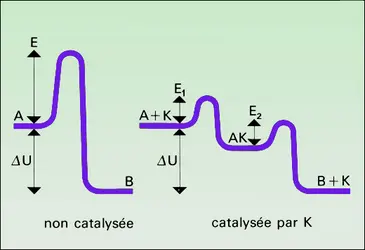
Diagramme énergétique
Encyclopædia Universalis France
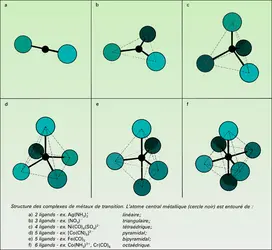
Métaux de transition : structure
Encyclopædia Universalis France
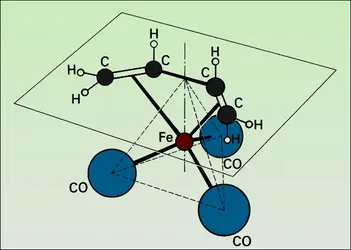
Fer carbonyle butadiène
Encyclopædia Universalis France
Autres références
-
ABZYMES
- Écrit par Joël CHOPINEAU , Encyclopædia Universalis , Alain FRIBOULET , Sabine PILLE et Daniel THOMAS
- 1 038 mots
Le concept d'anticorps catalytique, ou abzyme (contraction d’antibodyet enzyme), fut énoncé dès les années 1940 par Linus Pauling. S’appuyant sur le fait que la réaction chimique de transformation d'une molécule en une autre passe par un état de transition, qui représente...
-
ACIDES & BASES
- Écrit par Yves GAUTIER et Pierre SOUCHAY
- 12 367 mots
- 7 médias
 Certaines réactions sont accélérées par la présence de substances étrangères (fréquemment il suffit de très faibles quantités), que l'on trouve inchangées en fin de réaction et que l'on nomme catalyseurs. Lorsqu'en solution le catalyseur est constitué par les ions H+ ou OH...
Certaines réactions sont accélérées par la présence de substances étrangères (fréquemment il suffit de très faibles quantités), que l'on trouve inchangées en fin de réaction et que l'on nomme catalyseurs. Lorsqu'en solution le catalyseur est constitué par les ions H+ ou OH... -
ANTIOXYGÈNES
- Écrit par Robert PANICO
- 2 341 mots
- 2 médias
 ...irradiation ultraviolette : l'absorption d'énergie lumineuse par la substance autoxydable facilite la rupture de la liaison R—H ; — par action de catalyseurs : les peroxydes organiques, en particulier le peroxyde de benzoyle, fournissent, par décomposition thermique, des radicaux...
...irradiation ultraviolette : l'absorption d'énergie lumineuse par la substance autoxydable facilite la rupture de la liaison R—H ; — par action de catalyseurs : les peroxydes organiques, en particulier le peroxyde de benzoyle, fournissent, par décomposition thermique, des radicaux... -
ARGILES
- Écrit par Daniel BEAUFORT et Maurice PAGEL
- 2 655 mots
- 7 médias
 L' origine de la vie pourrait se trouverdans les propriétés catalytiques des surfaces des minéraux argileux qui auraient favorisé la polymérisation de molécules organiques complexes capables de donner naissance à des structures moléculaires aussi complexes que l'ARN. Les phyllosilicates...
L' origine de la vie pourrait se trouverdans les propriétés catalytiques des surfaces des minéraux argileux qui auraient favorisé la polymérisation de molécules organiques complexes capables de donner naissance à des structures moléculaires aussi complexes que l'ARN. Les phyllosilicates... - Afficher les 24 références
Voir aussi
- CARBONYLES MÉTALLIQUES
- CARBÈNES
- POLYMÉRISATION
- ÉTHYLÈNE
- ENZYMES
- HYDROLYSE
- RÉACTIONNELS MÉCANISMES
- BASE, chimie
- CATALYSE ENZYMATIQUE
- CATALYSE HOMOGÈNE
- CATALYSE HÉTÉROGÈNE
- HAMMETT FONCTION DE
- INHIBITEURS, chimie
- FORCE D'UN ACIDE ou D'UNE BASE
- ÉQUILIBRE, chimie
- CATALYSEURS
- HYDROXYLE ou OXHYDRYLE
- VITESSE DE RÉACTION
- CHIMISORPTION
- LIGAND ou COORDINAT, chimie
- FRIEDEL & CRAFTS CATALYSEUR DE
- HYDROGÉNATION
- ACTIVATION, chimie
- CENTRE ACTIF, chimie
- AIRE SPÉCIFIQUE
- AUTO-INHIBITION, chimie
- THERMODYNAMIQUE CHIMIQUE
- PROMOTEUR, chimie
- OXYDATION
- INITIATEUR, chimie
- FRITTAGE
- NUCLÉOPHILES SUBSTITUTIONS
- PLATINE
- SOLIDES PHYSIQUE DES
- SAPONIFICATION
- AMMONIUM QUATERNAIRE
- TRANSITION MÉTAUX DE
- OXYDES
- MONOXYDE DE CARBONE ou OXYDE DE CARBONE (CO)
- RÉACTIVITÉ CHIMIQUE
- ACIDITÉ ÉCHELLE D'
- SÉLECTIVITÉ, chimie
- CATALYSE TRANSFERT DE PHASE PAR
