ORGANOMÉTALLIQUES COMPOSÉS
Article modifié le
Organomagnésiens
Le réactif de Grignard
V. Grignard constata vers 1900 qu'une solution de bromure d'éthyle dans l'éther anhydre dissout des copeaux de magnésium de pureté suffisante par une réaction exothermique qui porte le solvant à une vive ébullition.
Le grand mérite de ce savant fut d'utiliser la solution ainsi préparée pour des synthèses sans chercher à en isoler les constituants. Cette découverte devait, dans les années qui suivirent, et encore aujourd'hui, fournir à la synthèse, tout au moins à l'échelle du laboratoire, l'un de ses plus précieux réactifs.

La réaction de Grignard a pu être étendue à d'autres dérivés halogénés : éthers chlorhydriques, bromhydriques, iodhydriques, bromure et iodure de phényle. Ce n'est que beaucoup plus tard qu'elle a pu être appliquée par l'emploi de solvants plus nucléophiles que l'éther (tétrahydrofuranne) aux halogéno-éthyléniques, aux halogéno-acétyléniques, au chlorure de phényle.
– La réaction de Grignard s'écrit schématiquement :
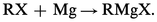
Ce n'est toutefois que l'expression globale d'une transformation, qui d'ailleurs n'est pas quantitative ; elle est plus ou moins accompagnée, comme il a été vu plus haut, de duplication ou de dismutation du radical R ; si R est saturé, le rendement augmente, des iodures aux bromures, puis aux chlorures, mais la facilité de l'attaque du métal varie en sens inverse. Les fluorures sont inactifs. En revanche, le rendement est meilleur avec les éthers halohydriques primaires qu'avec les secondaires ou les tertiaires, et, dans le cas des halogénures α-éthyléniques, ce n'est qu'au prix de sévères précautions qu'on évite en partie des duplications.

Réactif de Grignard
Encyclopædia Universalis France
De nombreuses hypothèses se sont succédé en ce qui concerne la nature des entités présentes dans le réactif de Grignard. L'une des plus généralement admises est l'existence d'un complexe solvaté (formule).
De fait, ce complexe a pu être isolé à l'état cristallisé, et l'analyse aux rayons X a confirmé la structure supposée. Mais des doutes subsistent sur la réalité de l'« équilibre de Schlenck » (réaction).
En effet, l'addition, au réactif de Grignard, d'une quantité suffisante de dioxanne provoque la précipitation de tous les halogènes de la solution sous forme de MgBr2 solvaté, et le liquide surnageant semble constitué du magnésien symétrique solvaté (R)2Mg, solvant.
L'évaporation laisse un résidu de ce solvat qui, sous vide poussé, abandonne son solvant et laisse subsister le magnésien symétrique (R)2Mg, poudre microcristalline spontanément inflammable et pratiquement inutilisable en synthèse.
Cependant, la nature des entités présentes dans le réactif de Grignard semble dépendre à la fois de celle de R, de celle du solvant et de celle de l'halogène, et aucune structure générale n'a pu s'imposer jusqu'ici.
Le comportement du réactif de Grignard est parfois le même que celui du magnésien symétrique solvaté, mais parfois il en diffère quantitativement et même qualitativement. C'est la raison pour laquelle les réactions qui vont être décrites sont celles que l'on observe avec les réactifs de Grignard bruts de fabrication, c'est-à-dire représentés globalement par la formule RMgBr, bien que les réactions accessoires laissent présumer que la teneur en halogénure de magnésium du milieu est toujours supérieure à la teneur indiquée par cette formule, et que cet excès de MgX2 modifie quelque peu les réactions observées avec un magnésien mixte « synthétique », c'est-à-dire résultant de l'addition stœchiométrique à (R)2Mg de MgX2, et d'un excès de solvant.
Les réactions de la solution de Grignard peuvent être classées en quatre groupes : action des composés à hydrogène mobile, action des composés à halogène mobile (ou à autre anion mobile), réactions d'addition, réactions utilisant plusieurs molécules d'organomagnésien mixte.
Schématiquement, tout se passe comme si l'halogénomagnésien mixte se dissociait ainsi :

Mais le solvant donne à la liaison R − Mg un caractère covalent suffisant pour différencier les magnésiens mixtes des dérivés sodés pour lesquels la dissociation totale :

Réaction des composés à hydrogène mobile
En principe, tous les composés dont le pKa est inférieur à 25 décomposent quantitativement les organomagnésiens :
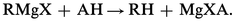
Cette échelle d'acidité suffisante s'étend des acides forts aux hydrocarbures acétyléniques vrais en passant par les acides faibles, les phénols, l' eau, les alcools, les amines non tertiaires. La nature de MgXA dépend de celle de A ; MgXA peut se comporter comme un sel métallique si A est un halogène, ou comme un complexe solvaté lorsque A = OR ; l'alcoolate magnésien mixte ROMgX est solvaté.
Dans le cas de l'eau, tout se passe comme si 2 MgXOH se décomposait en MgX2 + Mg(OH)2. Dans celui des carbures acétyléniques vrais, le dérivé R − C ≡ C − MgX, solvaté, est un nouvel organométallique.
Seule cette dernière réaction présente un intérêt en synthèse ; les autres ne constituent qu'une préparation de l'hydrocarbure RH à partir de RMgX, donc, initialement, de RX (ou de ROH). Bien entendu, l'eau est le réactif le plus indiqué pour réaliser cette protolyse, mais, afin d'éviter un empâtement du milieu par la magnésie précipitée, il est plus intéressant de faire appel à une solution acide diluée qui solubilise cette magnésie et rend l'extraction de l'hydrocarbure plus aisée ; c'est évidemment inutile si cet hydrocarbure est plus volatil que l'éther (préparation du méthane ou de l'éthane), mais il convient de débarrasser le gaz de l'éther entraîné, par barbotage dans l'acide sulfurique, ou de substituer à l'éther un éther-oxyde moins volatil (oxyde de butyle). Le même changement de solvant s'impose si l'hydrocarbure cherché a une volatilité voisine de celle de l'éther.
Réactions de composés à anion mobile
Halogènes et dérivés halogénés
Les halogènes agissent principalement dans le sens :

Cette réaction permet, par exemple, de passer de RBr à RI ; elle est employée pour le dosage de l'organomagnésien (cf. infra).
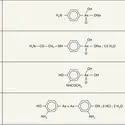
Les halogénures métalloïdiques, apparentés aux halogénures d'acide, les halogénures des métaux de transition plurivalents décomposent les organomagnésiens ; voici quelques exemples :

Le produit obtenu n'étant pas solvaté, le résultat est simple. Il se complique dans le cas des halogénures de métaux lourds : SnCl4, selon les proportions, conduit à RSnCl3, à (R)2SnCl2, à (R)3SnCl et à Sn(R)4 ; HgCl2 conduit à RHgCl ou à (R)2Hg.
Les choses se compliquent encore si le produit résultant est solvaté. La même ambiguïté se présente pour la composition du liquide obtenu que pour le réactif de Grignard : il est en effet difficile de savoir si AlCl3 conduit à RAlCl2, à (R)2AlCl et à (R)3Al solvatés ou à leur mélange en équilibre ; il en est de même pour ZnCl2 ou CdCl2.
Parmi les dérivés halogénés organiques, seuls les chlorures d'acides agissent régulièrement (cf. infra).

Réactions de composés à anion mobile
Encyclopædia Universalis France
Les halogéno-éthyléniques, les halogénures phényliques et la plupart des éthers halohydriques non tertiaires sont sans action en l'absence de catalyseurs. Mais, sous l'action du chlorure de cobalt anhydre, on observe une réaction radicalaire se traduisant par une soudure ou, plus souvent, par une dismutation (réaction).
Par contre, les halogénures α-non saturés donnent lieu, sans catalyseur, à une condensation (réaction).
Avec les homologues, on observe une transposition partielle qui est prépondérante avec les halogénures propargyliques (réaction).
Les iodures et bromures tertiaires agissent facilement (réaction). Plus réactifs, les éthers sulfuriques, même primaires, conduisent à une condensation analogue.
Enfin, l'éther chlorométhylique se condense sans catalyseur.
Groupes OR et éthers minéraux
Le groupe éthoxyle est bien moins actif que le groupe halogénure. Les éthers oxydes et les acétals n'agissent qu'exceptionnellement ; au contraire, les ortho-éthers et l'éther orthocarbonique conduisent à des acétals (réaction) ou à des cétals.
Les esters agissent comme les chlorures d'acides.
Réactions d'addition
L'oxygène s'additionne aux organomagnésiens :
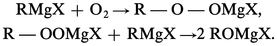
À l' hydrolyse, on recueille l'hydroperoxyde R − OOH (ou C6H5 − OOH) ou l' alcool ROH (ou le phénol C6H5 − OH).
Le soufre se comporte comme l'oxygène.
Les réactions les plus intéressantes sont les additions sur une liaison multiple entre carbone et hétéroatome ; il faut signaler ici que l'addition sur une liaison multiple carbone-carbone n'est que tout à fait exceptionnelle.
Le dioxyde de carbone conduit à un carboxylate magnésien mixte (carbonation) :

Le carboxylate est détruit par les acides minéraux dilués (synthèse des acides carboxyliques) :

Toutefois, la condensation avec d'autres molécules RMgX peut conduire à des cétones ou à des alcools tertiaires.
Les dérivés carbonylés conduisent à des alcoolates et, après hydrolyse, aux alcools :

C'est la synthèse la plus générale des alcools, primaires si l'on part de l'aldéhyde formique, secondaires dans le cas de tous les autres aldéhydes, tertiaires dans celui des cétones ; cependant, si la cétone est α-éthylénique, l'addition peut se faire plus ou moins partiellement en -1,4 sur le système conjugué (réaction).
À l'hydrolyse, on isole l'énol correspondant qui se transforme spontanément en cétone ; on assiste donc à une « addition apparente » sur la double liaison éthylénique.
De plus, si la cétone est encombrée et si l'organomagnésien porte de l'hydrogène sur le carbone en α, l'addition est concurrencée par une réduction (réaction).
À l'hydrolyse, on aboutit alors à un alcool secondaire. Enfin, une partie du magnésien peut énoliser la cétone, et à l'hydrolyse on retrouve une partie de la cétone mise en œuvre.
Les nitriles donnent lieu à une addition semblable :

Si le nitrile possède un hydrogène acide, cette réaction est éclipsée par celle conduisant à un éniminure qui à l'hydrolyse redonne le nitrile initial : il n'y a plus de synthèse.
Les imines stables agissent comme les cétones :

L'hydrolyse conduit à l'amine :

Réactions utilisant plusieurs molécules d'organomagnésien
Dans une première phase, les chlorures d'acides et les esters forment vraisemblablement un composé d'addition :

On ne sait si ces complexes se décomposent en cétone R − CO − R′ et, respectivement, en MgXCl ou en C2H5OMgX. Toutefois, la cétone apparaît après hydrolyse ; l'arrêt à ce stade exige une température très basse (− 80 0C) et l'« addition inverse » (on fait couler l'organométallique dans la solution éthérée du chlorure d'acide ou de l'ester). À la température ordinaire et par addition directe, on aboutit à un alcoolate tertiaire ; les esters formiques, toutefois, conduisent à un alcool secondaire.
On ignore si cette seconde réaction est due à l'attaque par une seconde molécule de RMgX du complexe d'addition ou de la cétone qui résulte de sa décomposition.
Les amides conduisent, en principe, à des cétones, mais, si l'amide n'est pas N-bisubstitué, la réaction consomme en pure perte une partie de l'organométallique, du fait de l'existence d'hydrogènes acides. La réaction n'est pratique que dans le cas du diéthylformamide :

L'hydrolyse libère l'amine (C2H5)2NH et l'aldéhyde R − CHO.
Les fonctions quadrivalentes : COCl2, ClCOOC2H5, CO(OC2H5)2, conduisent par des intermédiaires supposés (chlorure d'acide, esters, cétones) à des alcoolates tertaires :

Dosage des organomagnésiens
Il est parfois utile de connaître la teneur du réactif de Grignard en organomagnésien, la préparation à partir de RX n'étant pas quantitative.
Deux méthodes sont employées :
– le dosage alcalimétrique de la magnésie libérée par l'hydrolyse :


Le thiosulfate dose l'iode non employé.
La première méthode est excédentaire, car elle dose comme organométalliques les produits d'une hydrolyse ou d'une oxydation accidentelle ; la seconde est déficitaire, car la réaction parasite :

Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Jacques METZGER : professeur de chimie organique à la faculté des sciences de Marseille
- Charles PRÉVOST : professeur à la faculté des sciences de Paris, à l'École centrale des arts et manufactures de Paris et à l'École normale supérieure de Fontenay-aux-Roses
Classification
Médias
Réactif de Grignard
Encyclopædia Universalis France
Réactions de composés à anion mobile
Encyclopædia Universalis France
Autres références
-
ALCYNES
- Écrit par Jacques METZGER
- 3 302 mots
- 5 médias
 Les organométalliques ainsi obtenus se comportent en général comme les organométalliques saturés, d'où leur emploi à l'introduction du groupe acétylénique dans de nombreuses synthèses.
Les organométalliques ainsi obtenus se comportent en général comme les organométalliques saturés, d'où leur emploi à l'introduction du groupe acétylénique dans de nombreuses synthèses. -
ALDÉHYDES ET CÉTONES
- Écrit par Jacques METZGER
- 7 356 mots
- 4 médias
 Lesorganométalliques sont également, au sens large, des réducteurs susceptibles de transformer des fonctions trivalentes en dérivés carbonylés. Les organolithiens et les organomagnésiens mixtes s'additionnent sur les nitriles en formant une imine, hydrolysée en cétone. Les chlorures d'acides réagissent...
Lesorganométalliques sont également, au sens large, des réducteurs susceptibles de transformer des fonctions trivalentes en dérivés carbonylés. Les organolithiens et les organomagnésiens mixtes s'additionnent sur les nitriles en formant une imine, hydrolysée en cétone. Les chlorures d'acides réagissent... -
ANTIMOINE
- Écrit par Encyclopædia Universalis et Jean PERROTEY
- 3 876 mots
- 3 médias
 ...très toxique, liquéfié dès − 18 0C, facilement décomposé par la chaleur et oxydable avec dépôt d'antimoine métallique et formation d'eau. Les dérivés alcoylés et arylés de la stibine sont, en règle générale, comparables à leurs homologues arséniés, mais moins stables et plus difficiles à...
...très toxique, liquéfié dès − 18 0C, facilement décomposé par la chaleur et oxydable avec dépôt d'antimoine métallique et formation d'eau. Les dérivés alcoylés et arylés de la stibine sont, en règle générale, comparables à leurs homologues arséniés, mais moins stables et plus difficiles à... -
ARSENIC
- Écrit par Jean PERROTEY
- 4 499 mots
- 2 médias
 ...aromatiques aux atomes d'hydrogène de l'arsine, puis, par halogénation ou oxydation, plusieurs milliers de composés arsenicaux organiques ont été préparés. Ils représentent la plus importante famille de composés organométalliques. Cela est dû à l'intérêt médical de ces produits et à l'importance de la...
...aromatiques aux atomes d'hydrogène de l'arsine, puis, par halogénation ou oxydation, plusieurs milliers de composés arsenicaux organiques ont été préparés. Ils représentent la plus importante famille de composés organométalliques. Cela est dû à l'intérêt médical de ces produits et à l'importance de la... - Afficher les 26 références
Voir aussi
- HYDROLYSE
- RÉACTIONNELS MÉCANISMES
- EAU, physico-chimie
- ORGANOMAGNÉSIENS COMPOSÉS
- ORGANOLITHIENS COMPOSÉS
- ORGANOMERCURIELS COMPOSÉS
- DOSAGE, chimie
- ALCALINS
- HALOGÉNÉS DÉRIVÉS
- CÉTONES
- ADDITION, chimie
- WURTZ MÉTHODE DE
- DISMUTATION, chimie
- ORGANOZINCIQUES COMPOSÉS
- GRIGNARD RÉACTION DE
- SCHLENCK ÉQUILIBRE DE
- REFORMATSKY RÉACTION DE
- HALOGÉNURES
