DÉTERMINATION DE LA STRUCTURE 3D DES PROTÉINES
Article modifié le
La cristallographie aux rayons X
Jusqu’au début des années 2020, la cristallographie aux rayons X (RX) a été la méthode reine. Son principe remonte à une expérience de Max von Laue qui, en 1911, a exposé un cristal de sulfate de cuivre (CuSO4) à un faisceau de rayons X et a observé une série de taches sur une plaque photographique placée perpendiculairement à l’axe du faisceau. Dans ce type d’expérience, les RX, qui sont diffusés par les électrons des atomes contenus dans le cristal, interfèrent entre eux et génèrent un spectre de diffraction. La distribution des taches de diffraction est révélatrice de la symétrie du cristal et leurs intensités sont fonction de la structure des molécules cristallisées. Cette technique est fondée sur le pouvoir amplificateur des interférences constructives des RX diffusés par une grande quantité de molécules empilées et orientées de façon périodique dans un cristal. Étant donné que les RX utilisés ont une longueur d’onde de l’ordre de 1 à 2 angströms (1 Å = 10–10 m), comparable au diamètre des atomes, ils interagissent avec les nuages électroniques de ces derniers. L’analogie est souvent faite entre le phénomène de diffraction des RX et la microscopie optique parce que, dans les deux cas, on se sert d’un rayonnement électromagnétique (RX ou lumière visible) pour grossir l’image d’un échantillon donné. Pourtant, cette analogie avec le microscope est un peu trompeuse parce que nous ne disposons pas de « lentilles » pour les RX, étant donné qu’ils sont dispersés par tout type de matière. On ne peut donc pas observer d’image directe, comme le permet le microscope optique, mais seulement mesurer les RX diffractés issus du cristal.
La diffraction n’est pas un phénomène physique évident à décrire. Les RX, dispersés dans toutes les directions, interfèrent mutuellement de façon soit destructive, soit constructive, ce qui génère des ondes ou « réflexions » qui sont mesurées sur un détecteur plat. On les appelle « réflexions » en référence à la loi de Bragg. En 1915, William Henry et William Lawrence Bragg (respectivement père et fils) proposent une méthode très visuelle pour la compréhension de la diffraction : ils ont défini une série de plans imaginaires qui traversent le cristal et qui se comportent comme des « miroirs » pour les RX. En réalité, ces plans ne fonctionnent comme des miroirs que pour un seul angle incident, d’où la nécessité de faire tourner le cristal dans le faisceau de rayons X pour mettre successivement tous les plans dans leur « bon angle de réflexion ». La densité électronique échantillonnée par chacun de ces plans est directement proportionnelle à l’intensité de la réflexion correspondante. C’est à ce niveau qu’une partie très importante de l’information nécessaire se trouve pour résoudre la structure de la macromolécule cristallisée. Le spectre de l’ensemble de ces « réflexions » reflète la symétrie interne du motif qui, en se répétant dans les trois directions de l’espace, constitue le cristal ; il est essentiel de déterminer cette symétrie pour pouvoir finaliser l’analyse.

Le fait d’enregistrer les « réflexions » sur un détecteur plan bidimensionnel élimine une part essentielle des données requises. En effet, les ondes diffractées portent une information qui les relie spatialement, à savoir leur phase. En termes mathématiques, ce qui est mesuré avec le détecteur correspond à la première transformée de Fourier (grosso modo, à la décomposition de l’image de l’objet en « réflexions »). Pour récupérer l’image originale, on doit prendre la racine carrée des intensités mesurées (les amplitudes) et les combiner avec les phases respectives. On appelle cette opération, qui additionne les amplitudes phasées, la deuxième transformée de Fourier. Étant donné que la transformée de la transformée régénère la fonction initiale, ce calcul donne une carte de la densité électronique correspondant à la molécule dans le cristal. Par comparaison, la lentille d’un microscope est ici « remplacée » par une « lentille » mathématique. Par la suite, on va se servir du graphisme moléculaire interactif sur ordinateur pour placer les atomes des acides aminés dans cette carte de densité et ainsi générer un modèle atomique en 3D de la protéine cristallisée. On devra, par la suite, minimiser les différences entre le spectre de diffraction calculé à partir des coordonnés atomiques de ce modèle et le spectre mesuré à partir du cristal, en modifiant et en complétant le modèle.
Dans le passage de la représentation bidimensionnelle des cartes de densité à la modélisation de la structure 3D d’une protéine, le problème de la détermination de la phase est le plus complexe en cristallographie aux rayons X, et sa résolution peut être très technique. Il peut être résolu en diffusant des complexes d’atomes lourds, comme le tétrachloroplatinate de potassium (K2PtCl4), qui vont se fixer dans le cristal, et en comparant, ensuite, le spectre de diffraction ainsi obtenu avec celui du cristal sans atome lourd. Dans le même but, on peut aussi, par biologie moléculaire, remplacer l’acide aminé méthionine de la protéine par de la sélénométhionine, ce qui ajoute un atome lourd facilement repérable, en des endroits bien précis de la protéine. Mais, désormais, puisqu’une grande partie des repliements protéiques sont déjà connus et répertoriés, on peut utiliser une technique, appelée remplacement moléculaire, qui permet de générer des phases à partir d’un modèle proche connu et correctement placé dans la maille cristalline de la protéine en cours d’étude.
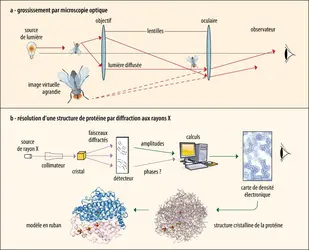
Détermination d’une structure 3D par diffraction des rayons X
carte de densité : P. Emsley et al./ University of California - CC-BY ; modèles : J.-C. Fontecilla-Camps/ Institut de biologie structurale ; adaptation : EUF
Malgré les aléas liés à la cristallisation (qui n’est pas reliée à la fonction biologique de ces macromolécules) et la perte d’information de la phase résultant de l’acquisition des données de diffraction sur un détecteur bidimensionnel, une grande partie des projets abordés par cristallographie ont été réussis. On entend souvent que cette technique est limitée par la qualité des cristaux, qui est fonction de la taille des molécules à analyser, et le fait que celles-ci soient figées par l’empilement cristallin. Pourtant, il s’est avéré que ces idées ne correspondent pas, dans la plupart des cas, à la réalité. Par exemple, la structure cristalline du ribosome (une organelle sur laquelle les protéines sont synthétisées), qui possède une masse de 2,5 millions de daltons (Da) et est composée de plusieurs protéines et acides nucléiques, a été déterminée en 1999 avec une bonne résolution. Comme les cristaux de protéines contiennent entre 40 % et 70 % de solvant, il est aussi possible de réaliser des études cinétiques in cristallo. Avec le développement du laser X à électron libre (XFEL) dans un laboratoire du CERN à Hambourg (Allemagne), il est devenu possible d’étudier simultanément des milliers de microcristaux dans la femtoseconde (10–15 s) et à température ambiante – dans le cas d’un seul cristal, il est normalement congelé pour éviter sa décomposition sous le faisceau de rayons X. Cette technique, qu’on appelle cristallographie aux RX sériée et qui utilise la « diffraction avant destruction », révolutionne les études cinétiques de la catalyse et la recherche de médicaments.
La cristallographie des protéines a connu des avancées techniques remarquables. Ainsi, on est passé de temps d’enregistrement des réflexions RX de plusieurs heures à seulement quelques minutes. Ce progrès est surtout lié aux gains en intensité du faisceau de RX des synchrotrons et à la sensibilité des détecteurs. Aussi, la collecte de données est très automatisée et la résolution des structures, très lourde en calcul, est devenue relativement rapide, grâce aux ordinateurs de plus en plus puissants et à des logiciels très performants.
Depuis 2020, une véritable révolution dans la prédiction de structures 3D de protéines s’est produite. Postérieurement, il en a été de même pour les acides nucléiques grâce à une méthode faisant appel à l’intelligence artificielle, appelée AlphaFold. Dans un premier temps, l’apport de cette approche à la cristallographie aux RX a surtout porté sur l’amélioration de modèles pour appliquer la technique de remplacement moléculaire évoquée plus haut à la résolution des structures dont on ne dispose pas de modèles expérimentaux assez proches.
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Emmanuelle NEUMANN : docteure en physique, ingénieure-chercheuse au CEA Grenoble
- Beate BERSCH : chargée de recherche au CNRS
- Juan FONTECILLA-CAMPS : conseiller scientifique au CEA
Classification
Médias
Détermination d’une structure 3D par diffraction des rayons X
carte de densité : P. Emsley et al./ University of California - CC-BY ; modèles : J.-C. Fontecilla-Camps/ Institut de biologie structurale ; adaptation : EUF
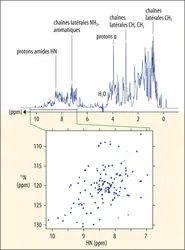
Spectres RMN 1D et 2D de protéine
Encyclopædia Universalis France
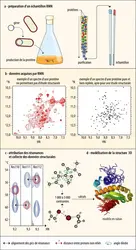
Détermination d’une structure tridimensionnelle par résonance magnétique nucléaire
images et conception : B Bersch/ Institut de biologie structurale ; adaptation : EUF
Voir aussi
- FRÉQUENCE, physique
- CONGÉLATION
- NOYAU ATOMIQUE
- MICROSCOPIE ÉLECTRONIQUE
- MICROSCOPIE ÉLECTRONIQUE À BALAYAGE EN TRANSMISSION
- INTERACTIONS MOLÉCULAIRES
- STRUCTURE, biologie
- RMN (résonance magnétique nucléaire), physico-chimie
- CRISTALLOGRAPHIE
- DIFFRACTION PAR LES CRISTAUX
- BIOLOGIE MOLÉCULAIRE
- HÉLICE, chimie des protéines
- LARMOR FRÉQUENCE DE
- MICROSCOPIE ÉLECTRONIQUE À BALAYAGE
- MOLÉCULES BIOLOGIQUES, structure et fonction
- MACROMOLÉCULES BIOLOGIQUES
- ISOTOPES, biologie
- TOMOGRAPHIE
- PROTÉINES
- BIOLOGIE HISTOIRE DE LA
- IMAGE ou REPRÉSENTATION TRIDIMENSIONNELLE (3D)
