DÉTERMINATION DE LA STRUCTURE 3D DES PROTÉINES
Article modifié le
La résonance magnétique nucléaire (RMN)
La résonance magnétique nucléaire a été découverte indépendamment par deux groupes constitués autour de Felix Bloch et Edward Purcell, dans les années 1945, aux États-Unis. La RMN est une méthode spectroscopique qui exploite une propriété magnétique de certains noyaux atomiques, le spin nucléaire, qui en est une propriété intrinsèque comme la charge ou la masse. Les caractéristiques magnétiques sont liées au nombre de protons ou neutrons que comporte le noyau, ainsi qu’au rapport gyromagnétique, une constante physique de base.
Principes de la méthode
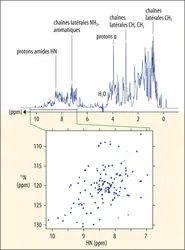
Spectres RMN 1D et 2D de protéine
Encyclopædia Universalis France

Lorsque des atomes possédant un spin nucléaire sont plongés dans un champ magnétique statique B0 très intense, les moments magnétiques des spins nucléaires, tels de petits aimants, prennent des orientations précises (quantifiées) et sont soumis à un mouvement de précession autour de l'axe du champ magnétique statique. Cette fréquence, aussi appelée fréquence de Larmor ou fréquence de résonance, est déterminée par le rapport gyromagnétique et par le champ magnétique local – c’est-à-dire l’intensité réelle du champ au niveau de chaque atome. En effet, le champ magnétique B0 polarise les nuages électroniques et induit ainsi des champs locaux qui s'ajoutent ou s'opposent à B0. Il en résulte que, dans une molécule donnée, différents noyaux ont des fréquences de Larmor qui diffèrent légèrement, en fonction de l'environnement chimique de chacun. Lors d'une expérience RMN, les fréquences de tous les atomes sont mesurées simultanément. Après l'application d’impulsions électromagnétiques générées par un champ oscillant à la fréquence de Larmor et perpendiculaire à B0, l'aimantation macroscopique résultant de l'ensemble de spins oscille dans le plan perpendiculaire au champ statique. Cette oscillation est détectée sous forme d'un courant généré par induction dans la bobine de détection : le signal de précession libre. Ce signal dans le domaine du temps est mathématiquement induit, par la transformée de Fourier, dans le domaine des fréquences. L’ensemble des fréquences ainsi déterminées constitue un spectre de signaux, le spectre RMN. Le déplacement chimique qui correspond à la fréquence de résonance (la position de chaque signal sur l'axe des fréquences exprimé en partie par million, ou ppm, par rapport à une référence) est le premier paramètre accessible. Par ailleurs, les spins peuvent communiquer entre eux : ces interactions spin-spin peuvent s'établir à travers des liaisons covalentes (interactions scalaires) ou à travers l'espace jusqu'à des distances de plusieurs angströms (interactions dipolaires).
Les expériences en RMN biomoléculaire utilisent des séquences d'impulsions adaptées à la manipulation des spins afin de pouvoir détecter ces interactions sous forme de changement d'intensité d'un signal donné ou, dans des expériences à multiples dimensions, sous forme de nouveaux signaux qui corrèlent ceux des noyaux dont les spins interagissent (pics de corrélation). En 2024, la librairie de séquences d'impulsions développée à l'Institut de biologie structurale à Grenoble pour la RMN biomoléculaire comporte ainsi plus de 120 séquences d'impulsions différentes. En général, la RMN biomoléculaire étudie les molécules en solution, ce qui permet la détection de spectres bien résolus avec des signaux fins. Bien qu’il soit aussi possible d'étudier les protéines à l'état solide (des cristaux, ou des protéines concentrées dans très peu d'eau), il est seulement question ici de la RMN biomoléculaire en solution.
Depuis les premières études de protéines par RMN dans les années 1960, des progrès technologiques majeurs ont contribué à la généralisation de l'utilisation de la RMN dans le domaine de la biologie structurale. Le développement d’aimants puissants (aimants supraconducteurs de plus de 20 teslas, avec des fréquences de Larmor des protons entre 600 et 1 200 MHz) a permis des gains significatifs en résolution et en sensibilité. L'utilisation de sondes de détection du signal refroidies à l'hélium ou à l'azote liquide, les cryosondes – l'électronique de la sonde et les préamplificateurs sont refroidis à des températures cryogéniques – réduit le bruit thermique et augmente significativement la sensibilité de la mesure. Enfin, parmi les éléments constituant les macromolécules biologiques, seuls le proton (1H, en RMN on utilise le terme « proton » plutôt que celui d’« hydrogène ») et le phosphore (31P) sont observables en abondance naturelle. Le développement des techniques de marquage par introduction d'isotopes 15N, 13C et 2H a permis d'étudier des protéines de tailles supérieures à 10 kDa. L'introduction uniforme des isotopes 15N et 13C est le prérequis pour la RMN multidimensionnelle moderne. Ce marquage facilite l'interprétation des spectres et réduit les ambiguïtés dans l'interprétation des données collectées. Le marquage au deutérium (2H) réduit la largeur des signaux observés et permet, souvent couplé à d'autres types de marquage très spécifiques, de travailler sur des protéines de taille supérieure à 25 kDa.
L'acquisition des données à partir d’une solution aqueuse de protéine purifiée se fait généralement à une concentration de quelques centaines de micromolaires (μmol/L) et à des températures comprises entre 5 et 40 °C. Elle peut prendre plusieurs jours, voire plusieurs semaines. La stabilité de la protéine est donc un facteur important.
Détermination des structures
En principe, le spectre RMN, c’est-à-dire l’ensemble des déplacements chimiques, contient toute l'information sur la conformation de la molécule. Mais le problème est trop complexe pour être résolu ab initio. Cependant, différents paramètres RMN apportent de l'information structurale, comme des distances ou des angles dièdres qui déterminent la conformation de la chaîne principale. La détermination d'une structure de macromolécule à partir des données RMN exige en général plusieurs étapes. Dans un premier temps, les résonances observées dans les spectres sont attribuées aux différents noyaux présents dans la protéine. Le marquage par des isotopes 15N et 13C visibles en RMN permet l'acquisition des spectres multidimensionnels, dans lesquels différents noyaux étudiés sont édités dans différentes dimensions. Leurs interactions (scalaires ou dipolaires) sont détectées sous forme de pics de corrélation. Ainsi, il est aisé d'attribuer les protons amides (HN), les azotes et les carbones de la chaîne principale en fonction de la séquence connue de la protéine. Une fois ces déplacements chimiques identifiés, leur analyse par des logiciels dédiés est effectuée pour prédire la conformation de la chaîne principale voire, pour des petites protéines, leur structure tridimensionnelle. Pour la détermination de structures à plus haute résolution, on mesure des paramètres expérimentaux supplémentaires reposant majoritairement sur l'effet Overhauser nucléaire (ou NOE pour Nuclear Overhauser Effect) qui est observé entre des protons proches dans l'espace, jusqu'à des distances de quelques angströms. Cela permet d’associer des protons à travers l'espace, et l'intensité des pics de corrélation est alors traduite en distance. Ainsi, on obtient typiquement 1 000 à 2 000 distances intramoléculaires qui sont utilisées comme contraintes dans le calcul de structure. Le principal défi est l'identification correcte des partenaires. En effet, il arrive fréquemment que plusieurs protons appartenant à des acides aminés différents dans la protéine aient le même déplacement chimique. La distance mesurée ne peut alors pas être attribuée à deux protons d'une manière certaine. Pour résoudre une partie des ambiguïtés, on utilise comme marqueur additionnel les 13C ou 15N auxquels les protons sont reliés. On peut également s'aider de modèles structuraux obtenus par la prédiction de structures 3D.
Le calcul de structure
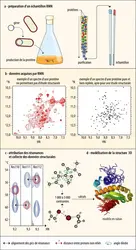
Détermination d’une structure tridimensionnelle par résonance magnétique nucléaire
images et conception : B Bersch/ Institut de biologie structurale ; adaptation : EUF
Il faut ensuite trouver l'arrangement spatial de tous les atomes de la protéine qui soit en accord avec l'ensemble des paramètres expérimentaux qui viennent d’être mesurés, mais aussi avec la géométrie connue des acides aminés qui composent la protéine (longueurs des liaisons entre acides aminés, angles entre ces liaisons, planéité des cycles aromatiques et de la liaison peptidique qui relie les acides aminés, etc.). Pour trouver cet arrangement, on utilise des simulations de dynamique moléculaire (in silico). Brièvement, lors d'une telle simulation, le modèle moléculaire de la protéine est chauffé à des températures très élevées, induisant des accélérations rapides des atomes, ce qui permet d'échantillonner l'espace conformationnel, la protéine pouvant adopter une grande variété de conformations différentes. Par la suite, les paramètres expérimentaux tels que les distances sont introduits sous forme de contraintes. Cela influencera les positions relatives des atomes et la chaîne de la protéine adoptera une conformation dans laquelle un certain nombre de ces contraintes seront respectées. Il n'y a pas de solution unique à ce problème, c'est pour cela qu'on répète cette simulation 200 à 1 000 fois, et le résultat, la structure de la protéine, est représenté sous forme d’un ensemble de structures (souvent 20) le plus en accord avec les contraintes expérimentales et géométriques.
Dynamique et interactions moléculaires
Parmi les trois techniques expérimentales de la biologie structurale, la RMN est la seule qui permet d'étudier des protéines intrinsèquement désordonnées ou comportant des séquences démunies d'une structure rigide. Grâce à l'analyse des déplacements chimiques, on peut détecter des éléments de structures secondaires, même formés de manière transitoire. La RMN permet également de déterminer la dynamique d'une protéine, qui est aussi reliée à sa fonction. La mobilité de la chaîne principale peut être quantifiée sur une large échelle de temps (allant de la picoseconde à la seconde). Son étude, par exemple à la suite d’une mutation, d’une interaction avec une protéine partenaire ou un ligand, permet une meilleure approche des aspects fonctionnels de la protéine.
Un autre atout majeur de la RMN est la détection et l'étude des interactions moléculaires. Le déplacement chimique est très sensible à l'environnement du noyau étudié. Une interaction avec une autre molécule induira des modifications des déplacements chimiques facilement détectables, ce qui permet d'identifier les sites d'interaction, information essentielle pour la modélisation de complexes entre molécules. De plus, la liaison, même transitoire, d'un petit ligand à une protéine de taille supérieure modifiera la mobilité du ligand. Ce phénomène peut être utilisé dans des criblages à haut débit de banques de molécules d’intérêt pharmacologique. La RMN est en effet une des rares techniques offrant la possibilité de détecter de faibles interactions. L'identification de motifs moléculaires simples capables d’interaction spécifique avec une protéine devient alors possible. Ces motifs moléculaires sont par la suite modifiés et éventuellement combinés afin de fournir des ligands spécifiques avec une forte affinité et éventuellement dotés d’une activité pharmacologique.
Polyvalente, la RMN permet de caractériser les propriétés structurales et dynamiques des protéines, d’obtenir des informations fonctionnelles par l'étude des interactions, soit avec d'autres protéines, soit avec des petites molécules ligands. Elle est de ce fait souvent utilisée en tant que technique complémentaire, apportant des informations au-delà de la structure tridimensionnelle, laquelle est souvent déterminée par une autre technique.
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Emmanuelle NEUMANN : docteure en physique, ingénieure-chercheuse au CEA Grenoble
- Beate BERSCH : chargée de recherche au CNRS
- Juan FONTECILLA-CAMPS : conseiller scientifique au CEA
Classification
Médias
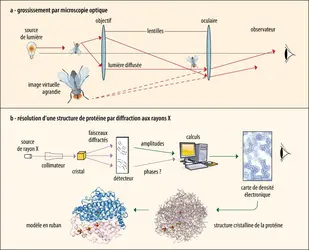
Détermination d’une structure 3D par diffraction des rayons X
carte de densité : P. Emsley et al./ University of California - CC-BY ; modèles : J.-C. Fontecilla-Camps/ Institut de biologie structurale ; adaptation : EUF
Spectres RMN 1D et 2D de protéine
Encyclopædia Universalis France
Détermination d’une structure tridimensionnelle par résonance magnétique nucléaire
images et conception : B Bersch/ Institut de biologie structurale ; adaptation : EUF
Voir aussi
- FRÉQUENCE, physique
- CONGÉLATION
- NOYAU ATOMIQUE
- MICROSCOPIE ÉLECTRONIQUE
- MICROSCOPIE ÉLECTRONIQUE À BALAYAGE EN TRANSMISSION
- INTERACTIONS MOLÉCULAIRES
- STRUCTURE, biologie
- RMN (résonance magnétique nucléaire), physico-chimie
- CRISTALLOGRAPHIE
- DIFFRACTION PAR LES CRISTAUX
- BIOLOGIE MOLÉCULAIRE
- HÉLICE, chimie des protéines
- LARMOR FRÉQUENCE DE
- MICROSCOPIE ÉLECTRONIQUE À BALAYAGE
- MOLÉCULES BIOLOGIQUES, structure et fonction
- MACROMOLÉCULES BIOLOGIQUES
- ISOTOPES, biologie
- TOMOGRAPHIE
- PROTÉINES
- BIOLOGIE HISTOIRE DE LA
- IMAGE ou REPRÉSENTATION TRIDIMENSIONNELLE (3D)
