
ÉDITION DES GÉNOMES
Article modifié le
Le ciblage de l’ADN : les méganucléases
Les limitations de la recombinaison homologue ont poussé à chercher des méthodes plus efficaces. En effet, sur un autre front de la recherche, les techniques de séquençage de l’ADN, qui permettent de connaître l’ordre d’enchaînement des nucléotides, ont progressé d’une manière fulgurante, de sorte que l’on dispose de plusieurs milliers de génomes complets d’organismes différents, allant de l’hydre d’eau douce au mammouth. S’il en est besoin, on séquence un génome humain (enchaînement de 3 milliards de nucléotides) en quelques jours. En se fondant sur des algorithmes bio-informatiques, on sait déterminer les séquences qui sont probablement des gènes. À partir de la seule séquence de l’ADN d’un génome, on déduit l’existence de plusieurs milliers ou dizaines de milliers de gènes, dont on ne connaît en général pas la fonction. Sur la base de la séquence codante de ces gènes, on peut seulement attribuer une fonction plausible aux protéines pour lesquelles ces gènes codent. Les conclusions sur les fonctions des gènes, ainsi obtenues par traitement mathématique des séquences d’ADN et extrapolation à partir des fonctions de protéines connues, font-elles sens sur le plan de la physiologie de l’organisme ? En d’autres termes, dire qu’un gène d’oursin code pour une protéine qui ressemble à une protéine impliquée dans le développement des mammifères ne dit pas qu’il s’agit d’un gène du développement de l’oursin. La seule manière de le savoir est d’inactiver ce gène en y introduisant une mutation, le réintroduire dans l’animal, puis en observer les effets sur le développement et la physiologie de l’organisme. Mais cette tâche est difficile car la capacité d’analyse fonctionnelle des gènes a progressé bien moins vite que la connaissance des génomes. De plus, elle est pratiquement impossible à réaliser dans la plupart des organismes, comme l’oursin de cet exemple, par manque des outils génétiques et moléculaires nécessaires.
Une méganucléase complexée à de l’ADN
Laguna Design/ SPL/ AKG-images
L’introduction de nouvelles technologies de modification rapide des génomes, connue sous le nom d’édition des génomes, est venue changer radicalement la situation depuis le début des années 2010. Une fois de plus, comme pour le génie génétique des années 1970, on s’appuie sur des « ciseaux moléculaires » que sont les enzymes de restriction qui coupent l’ADN au niveau de séquences particulières. Certaines de ces enzymes, comme I-Sce1 découverte en 1992 dans les mitochondries de la levure, reconnaissent une séquence non pas de six nucléotides (valeur moyenne pour la majorité des enzymes de restriction) mais d’au moins 18 nucléotides. Ces enzymes sont appelées méganucléases. Le préfixe « méga » est utilisé en référence à la longueur de la séquence ADN reconnue tandis que « nucléase » renvoie à l’activité de coupure de l’ADN (activité endonucléase). Du fait de sa taille, cette longue séquence ne se retrouve, en général et au mieux, qu’une fois dans un génome : on ne trouve la séquence reconnue par I-Sce1 statistiquement qu’une fois pour 15 à 20 milliards de nucléotides. La coupure au niveau de ce site unique peut ensuite être utilisée pour créer une modification locale de l’ADN, donc une mutation, qui peut inactiver le gène ou le modifier, ce qui permet d’étudier sa fonction ou de corriger une anomalie. La coupure peut également servir à intégrer un autre gène. Cette enzyme I-Sce1 a été utilisée en 1999 pour introduire une mutation ciblée unique chez la drosophile.
L’usage de telles enzymes pour la modification programmée des génomes est clairement formulé dès cette époque. Cependant, les méganucléases naturelles sont d’usage limité. La principale difficulté concerne le nombre limité de sites de reconnaissance, c’est-à-dire de séquences d’ADN reconnues. Certes, on connaît[...]
La suite de cet article est accessible aux abonnés
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Gilles SAUCLIÈRES : journaliste scientifique
Classification
Médias
Une méganucléase complexée à de l’ADN
Laguna Design/ SPL/ AKG-images
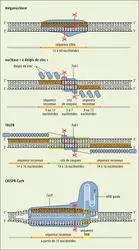
Modes d’action des outils d’édition du génome
Encyclopædia Universalis France

Emmanuelle Charpentier et Jennifer Doudna
Miguel Riopa/ AFP
