GÉNOMIQUE : ANNOTATION DES GÉNOMES
Article modifié le
Reconstruire une séquence d’ADN génomique complète
Un génome n’est jamais séquencé en une seule fois : les séquenceurs ne produisent que des fragments de courte taille par rapport à celle du génome. Ceux de première génération permettent de décrypter la séquence d’un fragment d’ADN, en une réaction, sur une longueur de 1 000 nucléotides au mieux ; les séquenceurs NGS permettent de séquencer en parallèle plusieurs millions de fragments différents longs de 35 à 700 paires de bases (pb) selon les technologies ; de nouveaux séquenceurs, dits de troisième génération, réalisent un séquençage en temps réel permettant l’obtention de séquences longues de plusieurs kilobases, mais ils souffrent d’un taux d’erreur important. Ainsi, pour avoir un génome entier, il faut dans un premier temps être capable de détecter les erreurs, d’ordonner et rabouter des fragments quelle que soit la méthode de séquençage utilisée.
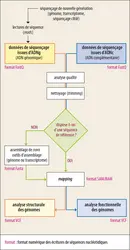
Étapes de l’analyse bio-informatique des séquences d’ADN
Encyclopædia Universalis France
Plusieurs millions de courtes séquences d’ADN sont donc obtenues à la fin du séquençage du génome d’un organisme. On les nomme « lectures » ou plutôt, comme en anglais,reads ; ils sont de longueur variable selon la technologie utilisée. Dans le cadre d’un séquençage de novo (pour une espèce dont le génome n’a encore jamais été déterminé), il faut ordonner ces lectures afin de reconstituer la séquence complète de l’ADN de chaque chromosome. Dans le cadre d’un reséquençage (pour une espèce dont le génome d’un individu, ou génome de référence, a déjà été déterminé), où les objectifs sont dirigés vers la connaissance des différences génétiques entre individus (polymorphismes), l’ordonnancement des lectures s’appuie sur la séquence déjà connue qui sert de référence. L’analyse bio-informatique qui permet d’aboutir à un génome complet se construit en plusieurs étapes. Dans tous les cas, on s’assure de la qualité des reads et de leur nettoyage puis on les assemble de novo si l’on ne dispose pas de génome de référence, ou on les aligne (mapping, en anglais) sur un génome de référence lorsqu’il existe.
Analyse de la qualité des reads
L’ensemble des reads est stocké, pour chaque individu, sous un format informatique particulier dans un fichier appelé FASTQ (« FASTA Quality »). Le séquenceur attribue, pendant l’analyse du signal généré par la lecture d’un nucléotide lors du séquençage de l’ADN, un score de qualité lié à la probabilité d’erreur d’identification de ce nucléotide, allant de 0 à 40. Un score de 10, 20 ou 30 correspond à une probabilité d’erreur, de 1/10, 1/100 ou 1/1 000 respectivement, dans l’identification d’un nucléotide donné à une position donnée. Ces scores sont encodés par un caractère unique, décrypté par le programme d’analyse qualité. Parfois, le séquenceur échoue à identifier un nucléotide, et il n’est pas rare de voir le symbole N (qui signifie « n’importe quel nucléotide ») dans la séquence d’un read, ce qui est associé à un mauvais score de qualité : on parle alors de bases ambiguës.
Le programme de référence pour l’analyse qualité des reads est donc l’outil FASTQC, développé par le Babraham Institute (Cambridge, Angleterre). Il permet de calculer et de visualiser graphiquement un ensemble de paramètres pour évaluer la qualité de l’ensemble des reads obtenus pour un individu.FASTQC propose notamment un graphique permettant de visualiser la distribution de qualité de chaque base pour chaque position dans le read. Cela est particulièrement utile au bio-informaticien qui pourra définir par la suite la taille de la région à retirer de l’analyse lors de l’étape suivante, en général des portions de séquence dont la qualité médiane est inférieure à 20. D’autres graphiques permettent de détecter l’existence de problèmes lors du séquençage comme le nombre de reads de trop faible score de qualité ou la présence de séquences surreprésentées.
Nettoyage des données (trimming)
Les reads retenus sont ensuite nettoyés : l’opération de nettoyage permet d’enrichir le jeu des données en reads de bonne qualité, en mesure d’être correctement assemblés ou comparés à un génome de référence. Dans le cadre d’un séquençage de novo, cette étape est essentielle pour diminuer le taux d’erreur et garantir un assemblage correct. Elle est réalisée grâce à des outils de nettoyage des données brutes de séquençage. Dans le cadre d’un reséquençage, le nettoyage sert surtout à enlever des nucléotides de mauvaise qualité qui pourraient être considérés, à tort, comme des variants de la séquence étudiée. Le seuil minimal de score de qualité est fixé à 20, mais les bio-informaticiens choisissent plutôt un score de 30. L’outil de nettoyage élimine les régions de faible score de qualité, généralement situées aux extrémités des reads. Cela concerne aussi les reads contaminés par une séquence adaptatrice introduite lors de la PCR, dont l’élimination – inutile dans le cas du reséquençage, car leurs séquences sont différentes de celles du génome analysé – est essentielle dans le cas d’un assemblage de novo puisque ces séquences conduiraient à des erreurs d’assemblage. Après le nettoyage, les readsdevenus trop courts – d’une taille inférieure à 20-25 nucléotides (nt) – sont éliminés, comme l’ont été ceux présentant une qualité globale faible (< 20 nt) ou présentant une proportion trop importante de nucléotides ambigus (> 50 %).
Assemblage de novo (sans génome de référence)
De manière imagée, cette étape consiste à résoudre un puzzle sans modèle. En effet, les lectures retenues sont finalement des petits fragments d'ADN qui, en se chevauchant partiellement, sont mis bout à bout, permettant de reconstruire de plus grands fragments d’ADN appelés contigs ; l'assemblage des contigs entre eux permet d'obtenir un scaffold ; les scaffolds assemblés entre eux aboutissent à un chromosome.L’assemblage de novo est très coûteux en termes de ressources mémoire et de temps de calcul, et nécessite des infrastructures bio-informatiques appropriées. Il implique a fortiori un nettoyage rigoureux des extrémités des reads afin de leur garantir une qualité élevée, puisque le principe de l’assemblage est basé sur le chevauchement entre les lectures.
On peut distinguer trois grandes familles d’algorithmes bio-informatiques d’assemblage existants :
Principe de l’approche dite Greedy pour l’assemblage de novo de fragments d’ADN
Encyclopædia Universalis France
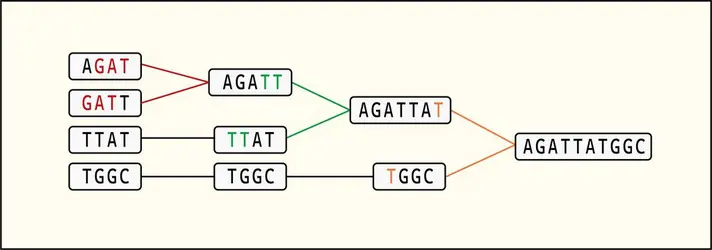
– La plus ancienne, l’approche dite Greedy (« glouton »), est fondée sur le calcul d’un score de chevauchement (plus ou moins grande identité des séquences entre paires de reads). Le couple présentant le score le plus élevé est fusionné en premier, donnant une séquence plus longue que chacun des reads. Puis le processus continue de proche en proche jusqu’à ce que chaque read soit associé à un autre et qu’apparaisse une seule séquence. Simple et intuitive, cette approche présente le désavantage d’être très gourmande en calcul (d’où son nom), et de connaître des difficultés à gérer les séquences répétées de faible complexité, nombreuses dans certains génomes, principales causes d’erreur dans l’assemblage.
Principe de l’approche OLC (Overlap Layout Consensus) pour l’assemblage de novo des produits de séquençage
Encyclopædia Universalis France
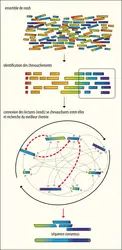
– Dans l’approche OLC (pour Overlap-Layout-Consensus, littéralement « chevauchement-mise en page-consensus »), la première étape vise à comparer les lectures entre elles et à identifier les paires chevauchantes. Une représentation graphique des chevauchements est construite au fur et à mesure avec les reads et les connexions entre deux reads s’il existe un chevauchement des séquences entre elles. L’algorithme cherche alors dans le graphe le chemin qui passe par tous les reads une fois et une seule. Cette seconde étape permet de déterminer l’ordre dans lequel les readsseront assemblés pour former un contig. La dernière étape vise à trouver une séquence consensus à partir des contigs obtenus précédemment.

Principe de l’approche de De Bruijn pour l’assemblage d’un génome
Encyclopædia Universalis France
– L’approche DBG (De Bruijn Graphs)est également fondée sur un graphe de chevauchement mais, avant son lancement proprement dit, le programme fragmente les reads en k-mers (toutes les combinaisons de sous-séquences chevauchantes de taille k), qui seront ensuite utilisées pour construire le graphe de De Bruijn. Les nœuds sont constitués par des éléments de taille k – 1 de chaque k-mer. Deux nœuds seront connectés entre eux s’ils partagent un chevauchement de taille k – 2. L’algorithme cherche ensuite le chemin qui passe par toutes les arêtes et une seule fois par arête. L’approche DBG est moins gourmande en calcul que les précédents et gère mieux les séquences répétées ou de faible complexité.

L’assemblage d’un génome de novo reste toutefois délicat malgré de nombreuses améliorations des algorithmes, notamment pour les grands génomes eucaryotes, présentant un contenu important en séquences nucléotidiques répétées. Les logiciels présentent chacun leurs spécificités propres et leur utilisation doit être en accord avec les données produites par le séquenceur. L’arrivée des séquenceurs de troisième génération, produisant de longs fragments, permet de faciliter les assemblages. Toutefois, ils produisent des données avec un fort taux d’erreurs (environ 10 %). De ce fait, on combine lors de l’assemblage, les lectures longues et les lectures courtes.
Alignement des lectures sur un génome de référence (mapping)
De manière imagée, l’étape dite de mapping consiste à résoudre un puzzle avec un modèle cette fois-ci. Les lectures sont simplement alignées sur un génome de référence, représentatif de l’espèce, puis raboutées. Les génomes de référence sont stockés dans des bases de données comme Ensembl, en Europe, NCBI (National Center for Biotechnology Information) ou UCSC (université de Californie à Santa Cruz) aux États-Unis, qui diffèrent quelque peu du point de vue de l’annotation génomique. On ne recherche pas ici un alignement parfait entre un read et la séquence de référence, car il est certain que l’on trouvera des différences, notamment dues à la variabilité entre individus.
Il existe une soixantaine d’outils d’alignement de lectures, qui diffèrent en fonction du type de reads à aligner (lectures courtes, lectures longues, lectures issues de séquençage d’ADNc ou d’ARN). La plupart fonctionnent en deux temps selon le principe du « chaînage et extension » : les readssont « découpés » en sous-ensembles de courtes séquences (k-mers) de tailles variables selon l’outil de mapping utilisé et (ou) le choix du bio-informaticien. Chaque k-mer sera recherché au long de la séquence du génome de référence. Lorsqu’une correspondance est trouvée entre la séquence du k-mer et une fraction de l’ADN connue (point d’ancrage-démarrage), l’outil cherche si la correspondance entre séquences peut être prolongée de part et d’autre, jusqu’à un maximum correspondant à la taille du read. Cette extension est prolongée tant que le score d’alignement augmente ; des mésappariements ou des « trous » (différences dues à des insertions ou délétions de nucléotides) sont toutefois autorisés tant que ces différences permettent de maximiser le score d’alignement. Pour chaque read aligné sur la séquence déjà connue, on trouvera une série d’informations, dont la position sur la séquence de référence, le nombre de nucléotides alignés et d’éventuelles insertions et délétions par rapport à la séquence de référence, les scores de qualité de l’alignement pour chaque nucléotide du read, dont on pourra déduire la probabilité que ce dernier soit aligné sur une mauvaise position.
Cas des séquences d’ARN messagers (RNA-Seq)
Les ARN messagers, copies des gènes actifs dans une cellule, ne sont pas séquençables directement, mais le deviennent après leur conversion en ADN complémentaire (ADNc). Le pool des ADNc donne des informations sur le « transcriptome » – c’est-à-dire l‘ensemble des gènes actifs – et sur le nombre de copies d’ARNm présentes dans une cellule (indication de l’activité du gène). Les premières étapes de traitement bio-informatique de ce type de données sont assemblables à celles de l’ADN génomique, avec cependant quelques particularités. L’analyse qualité de détection des biais techniques sera interprétée différemment. Le nettoyage des données devra tenir compte en particulier des séquences surreprésentées dues aux gènes fortement exprimés et de produits d’amplification par PCR qui ne sont pas artéfactuels et doivent être conservés.
Après nettoyage des données, trois situations se rencontrent :
– Si l’on ne dispose d’aucune séquence de référence (génome ou transcriptome), des outils d’assemblage de novo permettent de reconstruire la séquence des différents ARNm d’origine. Tout comme les logiciels d’assemblage de génome, ces outils requièrent de grandes ressources mémoire et un temps de calcul important, et surtout des reads d’excellente qualité.
– S’il existe une séquence génomique de référence, on utilisera un outil de mapping pour identifier la position initiale dans le génome de chacun des reads issus du RNA-Seq.
– Si l’on dispose d’un transcriptome de référence, on peut utiliser les mêmes outils que pour l’ADN génomique, mais cette méthode, certes très rapide, n’est pas la plus utilisée car on n’est jamais vraiment sûr de disposer d’un « vrai » transcriptome de référence.
En fait, l’alignement sur un génome de référence est préféré. Mais, dans ce cas, les outils de mapping sont différents pour les eucaryotes du fait de la structure en exons/introns de leurs gènes : ils doivent donc être capables de reconnaître un read à cheval sur deux exons.Or, deux régions exoniques peuvent être séparées dans le génome par un intron long de centaines voire de milliers de nucléotides : ainsi dans le gène de la dystrophine long de 2,4 millions de nucléotides, les exons (donc transcrits) ne représentent que 14 000 nucléotides. Par cette méthode, on pourra découvrir de nouveaux transcrits (long ARN non codant, par exemple), de nouveaux variants d’épissage (type de raboutage entre exons) et de nouvelles structures de gènes.
La suite de l’analyse des données de RNA-Seq dépend des objectifs expérimentaux. Souvent, cette technique est utilisée pour quantifier l’expression des gènes, via une comparaison entre différentes conditions expérimentales (pathologique versus sain, traité versus non traité, muté versus non muté, etc.) : on parle alors d’analyse d’expression différentielle. On réalise un comptage du nombre de reads obtenus pour chaque gène, sous l’hypothèse que ce nombre est proportionnel à l’abondance de l’ARN dans la cellule. Ce comptage peut se faire au niveau des transcrits complets, ou au niveau des exons, ce qui est plus utilisé dans une analyse différentielle de l’épissage alternatif. Après le comptage, une étape de normalisation (visant à rendre les échantillons comparables entre eux) devra être réalisée.
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Véronique BLANQUET : professeure de génétique, université de Limoges
- Stéphanie DURAND : maître de conférences, faculté des sciences et techniques de Limoges
Classification
Médias
Étapes de l’analyse bio-informatique des séquences d’ADN
Encyclopædia Universalis France
Principe de l’approche dite Greedy pour l’assemblage de novo de fragments d’ADN
Encyclopædia Universalis France
Principe de l’approche OLC (Overlap Layout Consensus) pour l’assemblage de novo des produits de séquençage
Encyclopædia Universalis France
Autres références
-
SÉQUENÇAGE DU GÉNOME HUMAIN, en bref
- Écrit par Nicolas CHEVASSUS-au-LOUIS et Encyclopædia Universalis
- 286 mots
Le 12 février 2001, les revues scientifiques Nature et Science publient la séquence quasi complète des trois milliards de bases du génome humain. Cette double publication conclut par un ex aequo la compétition entre un consortium international de laboratoires publics, qui a commencé ses...
-
BIOTECHNOLOGIES
- Écrit par Pierre TAMBOURIN
- 5 368 mots
- 4 médias
 Des gènes responsables de la résistance aux herbicides, aux infections virales, fongiques ou bactériennes, identifiés grâce au progrès de lagénomique, sont utilisés pour transférer aux plantes ces mêmes propriétés de résistance. C'est en 1985 que les premiers essais en champ de plantes transgéniques...
Des gènes responsables de la résistance aux herbicides, aux infections virales, fongiques ou bactériennes, identifiés grâce au progrès de lagénomique, sont utilisés pour transférer aux plantes ces mêmes propriétés de résistance. C'est en 1985 que les premiers essais en champ de plantes transgéniques... -
BOTANIQUE
- Écrit par Sophie NADOT et Hervé SAUQUET
- 5 647 mots
- 7 médias
 ...recherche scientifique s'est accélérée, engendrant des bouleversements imprévisibles de notre compréhension du monde, y compris celui des plantes. La génomique, par exemple, est en train de révéler une évolution et un fonctionnement bien plus complexe des génomes d'eucaryotes (organismes pourvus d'un...
...recherche scientifique s'est accélérée, engendrant des bouleversements imprévisibles de notre compréhension du monde, y compris celui des plantes. La génomique, par exemple, est en train de révéler une évolution et un fonctionnement bien plus complexe des génomes d'eucaryotes (organismes pourvus d'un... -
CANCER - Cancer et santé publique
- Écrit par Maurice TUBIANA
- 14 762 mots
- 8 médias
 ... représente un autre domaine de recherche. Maintenant qu'ont été identifiés les défauts du génome caractérisant les cellules cancéreuses, il est tentant d'essayer de les corriger et de faire redevenir normales les cellules cancéreuses. Quelques résultats ont été obtenus chez l'homme dans...
... représente un autre domaine de recherche. Maintenant qu'ont été identifiés les défauts du génome caractérisant les cellules cancéreuses, il est tentant d'essayer de les corriger et de faire redevenir normales les cellules cancéreuses. Quelques résultats ont été obtenus chez l'homme dans... - Afficher les 29 références
Voir aussi
- NUCLÉOTIDES
- GÉNOTYPE
- CODON, biologie moléculaire
- ARN MESSAGER ou ARNm
- EUCARYOTES
- SÉQUENÇAGE, génétique moléculaire
- GÉNOME
- THÉRAPIE GÉNIQUE
- BIOLOGIE MOLÉCULAIRE
- BANQUE DE DONNÉES
- CHROMOSOMES
- EXPRESSION GÉNÉTIQUE
- EXON
- INTRON
- NUCLÉOTIDIQUE SÉQUENCE
- GÉNÉTIQUE MOLÉCULAIRE
- TECHNIQUES HISTOIRE DES, XXe et XXIe s.
- PROTÉINES
- ADN COMPLÉMENTAIRE ou ADNc
