
GÉNOMIQUE : ANNOTATION DES GÉNOMES
Article modifié le
Annotation de la séquence d’ADN génomique
L’établissement de la séquence d’un génome n’est que la première étape dans la compréhension de son fonctionnement. Cette étape longtemps limitante, ne l’est plus du fait du développement du NGS, même si elle requiert une infrastructure et un personnel bio-informatique compétent dans la reconstruction du puzzle génomique. À son issue, on dispose d’une succession de nucléotides A, T, C et G, dépourvue de sens évident. L’étape suivante consiste donc en l’annotation du génome, qui se subdivise en deux niveaux :
– une annotation structurale visant à identifier les zones fonctionnelles du génome (par exemple, les gènes) et les éléments régulateurs de cette fonctionnalité (par exemple, les promoteurs ou toute autre séquence régulatrice) ;
– une annotation fonctionnelle visant à déterminer la ou les fonctions des gènes.
Identifier les gènes
L’annotation structurale s’appuie sur deux approches complémentaires :
– une approche ab initio (ou méthode intrinsèque) fondée sur la recherche d’éléments de séquence caractéristiques des gènes via l’utilisation d’outils de prédiction de ces derniers ;
– une approche comparative (ou méthode extrinsèque) fondée sur la recherche de similarité entre les séquences étudiées et les séquences existantes, connues, disponibles dans les bases de données.
Recherche de séquences potentiellement codantes
Si l’on cherche à identifier les séquences pouvant coder une protéine, la stratégie la plus simple est la méthode dite d’ORFing (pour ORF Finding) qui s’appuie sur la recherche de phase ouverte de lecture (ORF, Open Reading Frame), c’est-à-dire la suite de triplets – ou codons, chacun codant pour un acide aminé) – codant pour la protéine la plus longue possible jusqu’au moment où l’on rencontre un codon stop (UAA, UAG ou UGA sur l’ARN) qui interrompt la traduction de l’ARN messager. Concrètement, l’outil effectue une traduction en acides aminés de la séquence nucléotidique dans les six cadres de lecture – trois cadres pour chaque brin (brin sens ou brin complémentaire dit antisens) – c’est-à-dire en décalant d’un nucléotide à chaque lecture à partir de l’extrémité 5’. En sélectionnant des ORF assez longues – il existe très peu de protéines de moins de 60 acides aminés – et celles commençant par un codon d’initiation de la traduction en protéine (AUG, par exemple), on peut identifier facilement les séquences potentiellement codantes qui signent la présence de gènes. Cette méthode, assez naïve, présente quelques limites : d’abord, les ORF identifiées ne correspondent pas nécessairement à des gènes... ; ensuite, elle n’est applicable qu’aux séquences d’ADN qui ne contiennent pas d’introns interrompant les séquences codantes donc, pour l’essentiel, celles du monde bactérien.
Les outils de prédiction des gènes utilisent une stratégie statistique entièrement différente, fondée sur le fait que le code génétique, tout universel qu’il soit, n’est pas utilisé de la même manière selon les organismes et les zones authentiquement codantes. Ainsi, certains codons synonymes (par exemple, codant le même acide aminé) sont utilisés préférentiellement chez certaines espèces et pas chez d’autres ; au sein d’une espèce, il existe aussi un usage du code génétique différent dans les espaces non codants. Finalement, les séquences codantes suivent l’usage du code de leur organisme d’origine, tandis que les séquences non codantes, non soumises à une pression de sélection, ne le suivent pas. Cette propriété statistique, qui conduit à un « style » de composition en nucléotides différent entre séquences codantes et non codantes, est exploitée par les outils de prédiction de gènes. L’algorithme de prédiction est construit à partir d’un ensemble de séquences « connues » (phase d’apprentissage)[...]
La suite de cet article est accessible aux abonnés
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Véronique BLANQUET : professeure de génétique, université de Limoges
- Stéphanie DURAND : maître de conférences, faculté des sciences et techniques de Limoges
Classification
Médias
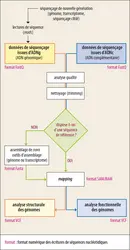
Étapes de l’analyse bio-informatique des séquences d’ADN
Encyclopædia Universalis France
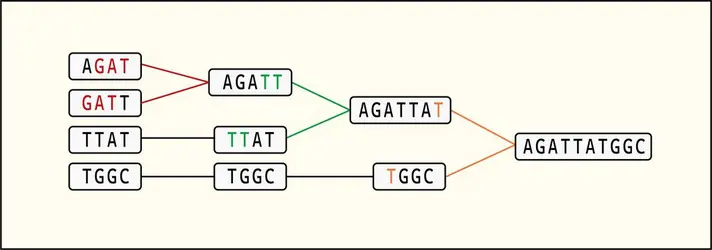
Principe de l’approche dite Greedy pour l’assemblage de novo de fragments d’ADN
Encyclopædia Universalis France
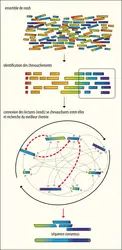
Principe de l’approche OLC (Overlap Layout Consensus) pour l’assemblage de novo des produits de séquençage
Encyclopædia Universalis France
Autres références
-
SÉQUENÇAGE DU GÉNOME HUMAIN, en bref
- Écrit par Nicolas CHEVASSUS-au-LOUIS et Encyclopædia Universalis
- 286 mots
Le 12 février 2001, les revues scientifiques Nature et Science publient la séquence quasi complète des trois milliards de bases du génome humain. Cette double publication conclut par un ex aequo la compétition entre un consortium international de laboratoires publics, qui a commencé ses...
-
BIOTECHNOLOGIES
- Écrit par Pierre TAMBOURIN
- 5 368 mots
- 4 médias
 Des gènes responsables de la résistance aux herbicides, aux infections virales, fongiques ou bactériennes, identifiés grâce au progrès de lagénomique, sont utilisés pour transférer aux plantes ces mêmes propriétés de résistance. C'est en 1985 que les premiers essais en champ de plantes transgéniques...
Des gènes responsables de la résistance aux herbicides, aux infections virales, fongiques ou bactériennes, identifiés grâce au progrès de lagénomique, sont utilisés pour transférer aux plantes ces mêmes propriétés de résistance. C'est en 1985 que les premiers essais en champ de plantes transgéniques... -
BOTANIQUE
- Écrit par Sophie NADOT et Hervé SAUQUET
- 5 647 mots
- 7 médias
 ...recherche scientifique s'est accélérée, engendrant des bouleversements imprévisibles de notre compréhension du monde, y compris celui des plantes. La génomique, par exemple, est en train de révéler une évolution et un fonctionnement bien plus complexe des génomes d'eucaryotes (organismes pourvus d'un...
...recherche scientifique s'est accélérée, engendrant des bouleversements imprévisibles de notre compréhension du monde, y compris celui des plantes. La génomique, par exemple, est en train de révéler une évolution et un fonctionnement bien plus complexe des génomes d'eucaryotes (organismes pourvus d'un... -
CANCER - Cancer et santé publique
- Écrit par Maurice TUBIANA
- 14 762 mots
- 8 médias
 ... représente un autre domaine de recherche. Maintenant qu'ont été identifiés les défauts du génome caractérisant les cellules cancéreuses, il est tentant d'essayer de les corriger et de faire redevenir normales les cellules cancéreuses. Quelques résultats ont été obtenus chez l'homme dans...
... représente un autre domaine de recherche. Maintenant qu'ont été identifiés les défauts du génome caractérisant les cellules cancéreuses, il est tentant d'essayer de les corriger et de faire redevenir normales les cellules cancéreuses. Quelques résultats ont été obtenus chez l'homme dans... - Afficher les 29 références
Voir aussi
- NUCLÉOTIDES
- GÉNOTYPE
- CODON, biologie moléculaire
- ARN MESSAGER ou ARNm
- EUCARYOTES
- SÉQUENÇAGE, génétique moléculaire
- GÉNOME
- THÉRAPIE GÉNIQUE
- BIOLOGIE MOLÉCULAIRE
- BANQUE DE DONNÉES
- CHROMOSOMES
- EXPRESSION GÉNÉTIQUE
- EXON
- INTRON
- NUCLÉOTIDIQUE SÉQUENCE
- GÉNÉTIQUE MOLÉCULAIRE
- TECHNIQUES HISTOIRE DES, XXe et XXIe s.
- PROTÉINES
- ADN COMPLÉMENTAIRE ou ADNc
