
HÉMOGLOBINOPATHIES
Article modifié le
Les maladies de l'hémoglobine
Les hémoglobines anormales sont responsables de maladies dont la cause est génétique et l'interprétation moléculaire, ce qui a permis à L. Pauling de parler de « maladie moléculaire ». Les anomalies observées sont de deux types : quantitatives et qualitatives.
Les anomalies quantitatives : thalassémies
Ce type d'anomalies est connu depuis longtemps sous le nom d' anémie de Cooley, ou thalassémie, ou anémie méditerranéenne. Cette anémie se rencontre souvent chez les peuples du bassin méditerranéen. En outre, elle peut prendre deux aspects : celui d'une anémie très grave avec hémolyse, hypochromie, globules rouges ayant l'aspect en cible, et anomalies grossières dans le taux des différentes hémoglobines (c'est le cas des sujets homozygotes) ; ou au contraire celui d'une anémie très modérée, avec peu de troubles (pour un sujet hétérozygote).
Cette maladie est due à l'absence ou à l'insuffisance de la synthèse d'un des types de chaînes de l'hémoglobine. On parle de β-thalassémie quand la synthèse de la chaîne β est affectée, d'α-thalassémie, quand celle de la chaîne α est affectée. Ces troubles ont été étudiés grâce aux méthodes de la génétique moléculaire, révélant l'absence de synthèse d'ARN normal : arrêt prématuré (codon stop intempestif) de la synthèse de la chaîne, délétions totales ou partielles du gène, anomalies dans l'excision-épissage, etc.
Les β-thalassémies
Les β-thalassémies sont très fréquentes, elles sont dues :
– soit à l'absence complète ou presque complète de synthèse de la chaîne β (β0-thalassémie ou thalassémie majeure) ; les troubles apparaissent dans la première année de la vie et la maladie est rapidement mortelle ; l'électrophorèse montre l'absence de Hb A1, la présence de Hb F, un taux élevé de Hb A2.
– soit à la synthèse partielle de chaîne β (β+-thalassémie ou thalassémie mineure). Les troubles dans ce cas sont bénins ; on retrouve un taux élevé de Hb A2 et la persistance de Hb F. Les taux sont variables d'une famille à l'autre, ce qui rend très hétérogène le tableau clinique, hématologique et biochimique.
Les α-thalassémies
Il peut s'agir soit du cas homozygote avec absence totale de synthèse de la chaîne α (α0-thalassémie), mortel au cours de l'évolution in utero ; soit du cas hétérozygote avec déficit partiel de la chaîne α (α+-thalassémie), assez bien toléré, car le gène normal assure une compensation. Dans ce dernier cas, on trouve un excès de chaînes β et γ par rapport à α. Cela se traduit par la présence, à côté des hémoglobines A1, A2 et F (synthétisées à faible taux), de formes anormales dues à l'association des chaînes normales β et γ en excès : Hb H (ou β4) et Barts (ou γ4).
Les anomalies qualitatives : hémoglobinoses
D'un point de vue médical, on distingue deux grands types d'hémoglobines anormales : certaines sont découvertes chez des sujets qui ne sont pas malades (par exemple au cours d'études systématiques de populations) ; d'autres provoquent des phénomènes pathologiques plus ou moins graves, par suite d'anomalies physico-chimiques ou fonctionnelles de l'hémoglobine. Seules ces dernières seront décrites ici.
Anomalies physico-chimiques
Anomalies de charge électrique
Elles sont loin d'être toujours nocives, et on n'insistera que sur la plus fréquente d'entre elles, l'hémoglobine S. Historiquement, sa découverte par L. Pauling et H. Itano a permis de relier pour la première fois une maladie à l'anomalie d'une molécule : c'est le premier et le meilleur exemple des « maladies moléculaires ».
Médicalement, son importance n'est pas moindre : à l'état homozygote, elle provoque une anémie hémolytique[...]
La suite de cet article est accessible aux abonnés
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Michel COHEN-SOLAL : maître de recherche à l'I.N.S.E.R.M., C.H.U. Henri-Mondor, Créteil
- Jean-Claude DREYFUS : professeur honoraire à la faculté de médecine Cochin-Port-Royal
Classification
Médias
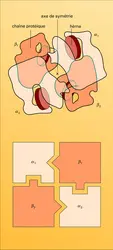
Hémoglobine : structure quaternaire
Encyclopædia Universalis France
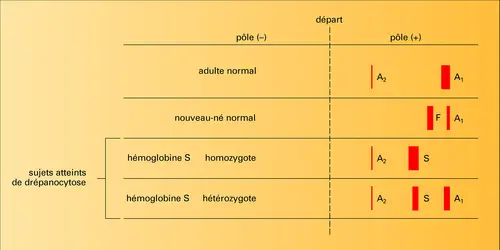
Hémoglobine : électrophorèse
Encyclopædia Universalis France
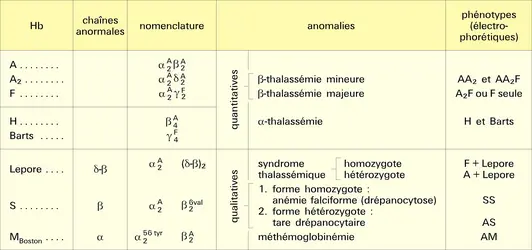
Hémoglobines et hémoglobinoses
Encyclopædia Universalis France
Autres références
-
PATHOLOGIE MOLÉCULAIRE DES HÉMOGLOBINES - (repères chronologiques)
- Écrit par Dominique LABIE
- 189 mots
1949 L. Pauling : isolement par électrophorèse de l'hémoglobine S de la drépanocytose.
1956 V. Ingram : notion de maladie moléculaire (différence de structure entre hémoglobines A et S).
1959 V. Ingram : phylogénie des hémoglobines et démembrement des thalassémies.
1960...
-
ANÉMIES
- Écrit par Bruno VARET
- 3 094 mots
- 5 médias
...– les anémies corpusculaires et constitutionnelles dues à une anomalie de la membrane ( sphérocytose héréditaire, elliptocytose héréditaire...), à une anomalie de l'hémoglobine sans défaut de synthèse (hémoglobine S homozygote, hémoglobine SC, hémoglobine instable) ; à une anomalie enzymatique (déficit... -
CYANOSE
- Écrit par François BOURNÉRIAS
- 145 mots
Coloration bleutée de la peau et des muqueuses, prédominant aux extrémités. Elle résulte d'une augmentation du taux de l'hémoglobine réduite dans le sang périphérique et apparaît lorsque ce taux dépasse 5 grammes pour 100 millilitres.
Il faut distinguer : la cyanose centrale...
-
FIÈVRE BILIEUSE HÉMOGLOBINURIQUE
- Écrit par François BOURNÉRIAS
- 223 mots
-
HÉMATOLOGIE
- Écrit par Jean BERNARD et Michel LEPORRIER
- 8 479 mots
- 1 média
 ...héréditaires, raciales ou familiales, qui atteignent des millions d'hommes en Afrique, en Asie, en Amérique, en Europe méditerranéenne sont dues à la présence d'hémoglobines anormales. Dans la structure de chaque hémoglobine se trouve une chaîne rigoureusement ordonnée d'une centaine d' acides aminés. Le changement...
...héréditaires, raciales ou familiales, qui atteignent des millions d'hommes en Afrique, en Asie, en Amérique, en Europe méditerranéenne sont dues à la présence d'hémoglobines anormales. Dans la structure de chaque hémoglobine se trouve une chaîne rigoureusement ordonnée d'une centaine d' acides aminés. Le changement... - Afficher les 14 références
Voir aussi
- MATURATION DE L'ARN
- HÉRÉDITAIRES MALADIES ou MALADIES GÉNÉTIQUES
- GLOBINE
- HÉMOGLOBINOSES
- TRANSCRIPTION, biologie moléculaire
- TRADUCTION, biologie moléculaire
- DRÉPANOCYTOSE ou ANÉMIE FALCIFORME
- MÉTHÉMOGLOBINE
- POLYPEPTIDIQUE CHAÎNE
- HÈME
- LEPORE HÉMOGLOBINE
- THALASSÉMIE
- HÉMATIE ou GLOBULE ROUGE ou ÉRYTHROCYTE
- CROSSING-OVER, génétique
- PROTÉINES BIOSYNTHÈSE DES
- HOMOZYGOTE
- HÉTÉROZYGOTE
- RÉCESSIVITÉ, génétique
- GÉNÉTIQUE MOLÉCULAIRE
- HÉMOPATHIES
- PROTÉINES
- ÉPISSAGE, génétique moléculaire
