HÉMOGLOBINOPATHIES
Article modifié le
Mécanismes génétiques de la production des hémoglobines anormales
Code génétique
Il est bien établi que la synthèse des protéines est sous la commande des gènes de structure, eux-mêmes faisant partie de l'acide désoxyribonucléique (ADN), c'est-à-dire du génome du noyau cellulaire. L'ADN est constitué d'une succession de groupements chimiques, des nucléotides, dont les éléments caractéristiques sont quatre bases : adénine (A), guanine (G), cytosine (C) et thymine (T). C'est l'agencement de ces quatre espèces moléculaires en séquences spécifiques qui constitue les gènes.
Comme tous les gènes des organismes pluricellulaires (eucaryotes), les gènes de globine comportent, au milieu de la séquence qui code l'information génétique nécessaire à la synthèse de la protéine, des séquences non codantes plus ou moins longues qui l'interrompent, les introns. Il existe deux introns par gène de globine, qui divisent les séquences codantes en trois zones ou exons.

Dans un premier temps, le gène dans son entier (exons et introns) est copié (transcription) en acide ribonucléique (ARN) qui lui est complémentaire, et dont les quatre bases sont A, G, C et uracile (U) qui remplace la thymine. Cet ARN précurseur va alors subir un certain nombre de réactions dans le noyau de la cellule, les principales consistant en l'excision des introns suivie de l' épissage des exons, ainsi que l'addition de bases méthylées du côté 5′ (cap) et d'une queue de plusieurs bases adénine (poly-A) du côté 3′. On retrouve alors dans le cytoplasme une molécule maturée d'ARN messager (ARNm).
C'est cet ARNm cytoplasmique qui va diriger alors la synthèse de la protéine ( traduction). Trois bases successives (un triplet) correspondent à un acide aminé. L'ensemble de ces triplets constitue le code génétique. Des signaux spécifiques conditionnent le bon déroulement de la traduction, tels que le codon d'initiation (AUG) et le codon de terminaison (UAA dans la plupart des gènes de globine humaine) qui contrôlent respectivement le début et la fin de l'incorporation des acides aminés dans la chaîne de globine.
Mécanisme des mutations
Une mutation est un événement qui se produit dans le gène de structure au niveau du génome et vient en modifier de manière définitive et plus ou moins importante la structure et de là retentit sur la protéine dont il commande la synthèse.
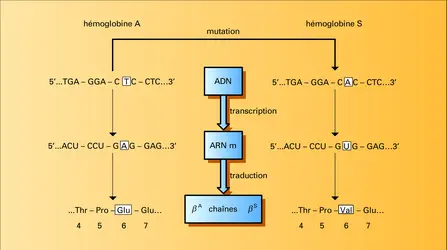
Mutation du gène de l'hémoglobine
Encyclopædia Universalis France
Une mutation ponctuelle consiste dans le changement d'une seule des bases d'un gène. Ce sont des mutations de ce type qui sont responsables le plus généralement des hémoglobines anormales. Cette erreur au niveau de l'ADN est transmise au cours de la transcription à l'ARNm, et par suite à la protéine synthétisée : le triplet modifié de l'ARNm correspond à un acide aminé autre que celui qui est normalement inseré dans la protéine. L'exemple de l'hémoglobine S permet de comprendre ce mécanisme.
Dans le cas de l'hémoglobine Lepore, le mécanisme est différent : il s'agit d'un crossing-over inégal entre les gènes δ et β dont la structure est assez semblable, mais qui sont localisés l'un à côté de l'autre sur le même chromosome.
On peut observer des cas de doubles hétérozygotes, c'est-à-dire des sujets hétérozygotes pour deux hémoglobines anormales différentes, ayant hérité d'une tare différente de chacun de leurs parents. Ces cas avaient permis de tirer des conclusions génétiques importantes, de nombreuses années avant que la génétique moléculaire moderne n'en apporte la confirmation :
– il n'y a aucune liaison génétique entre les gènes des chaînes α et β qui sont portés par deux paires de chromosomes différents, tandis que ceux des chaînes β et δ sont étroitement liés sur le même chromosome ; il en est de même d'ailleurs pour les gènes de la chaîne γ ;
– il existe plus d'un gène pour la chaîne α, car on a pu observer des sujets doubles hétérozygotes produisant trois types de chaînes α (deux chaînes α anormales en plus de la chaîne α normale) ;
– par contre, les doubles hétérozygotes pour deux hémoglobines anormales dont l'anomalie porte sur la chaîne β ne fabriquent pas du tout d'hémoglobine A normale. Le locus génétique de ces hémoglobines anormales est le même que celui de l'hémoglobine normale ; on dit qu'il est allélomorphe, ce qui veut dire que ce locus génétique ne peut former qu'un seul type de chaîne, soit normal, soit porteur d'une anomalie déterminée. Ainsi les individus portant simultanément l'Hb S et l'Hb C ont pour génotype α2A/βSβC.
Les lésions moléculaires des thalassémies sont d'élucidation beaucoup plus récente. Il a fallu attendre que l'on sache isoler les gènes par les méthodes du génie génétique dans la fin des années 1970, et que leur séquence ait été établie. Les lésions perturbant ou empêchant l'expression normale du gène, principalement au niveau de ses signaux spécifiques, peuvent bloquer la maturation de l'ARN précurseur en ARNm, et partant empêcher toute synthèse protéique normale. Des mutations ponctuelles ou des pertes de quelques bases (délétions) à la jonction intron-exon empêchent le phénomène d'excision- épissage, donc arrêtent la production d'ARNm mûr et sont responsables de thalassémies β0, ou α0 plus rarement. Par ailleurs, des mutations faisant apparaître une séquence semblable à une jonction intron-exon à un endroit quelconque peuvent tromper le système de maturation. Comme le signal normal est toujours présent, le phénomène d'excision-épissage peut se produire à deux endroits différents : d'une part au site normal, aboutissant à un ARNm mature normal, donc commandant la synthèse d'une protéine normale, mais en quantité réduite, et d'autre part au nouveau site, aboutissant à un ARNm aberrant. Un tel phénomène est responsable des β+-thalassémies, entre autres.
Un certain nombre de défauts traductionnels ont été décrits dans les thalassémies. Un codon non-sens nouveau, consécutif à une mutation ponctuelle, va interrompre prématurément la synthèse protéique. Cela conduira à un génotype de β0-thalassémie ; on le rencontre de manière assez fréquente dans les populations méditerranéennes (β0 39, car le nouveau codon non-sens est en position 39 de la chaîne β) ou asiatiques (β0 17). Un autre type de mutation consiste en la délétion d'une base, ce qui décale la phase de la lecture du message, et conduit à la synthèse d'une protéine aberrante et à l'absence de chaîne normale.
Enfin, des délétions plus ou moins larges des gènes de structure vont empêcher la synthèse de tout ARNm, donc de la chaîne protéique correspondante. De telles disparitions de gènes ont été principalement observées dans les α0-thalassémies. Cependant, l'existence de plusieurs gènes α sur le génome humain rend les phénotypes assez hétérogènes en fonction du nombre de gènes emportés par la délétion. Des délétions spécifiques du gène β responsable de β0-thalassémie n'ont été que très rarement montrées. Par contre, des délétions d'une très grande portion de génome englobant les gènes δ et β ont été décrites dans des formes graves dites δ0β0-thalassémies, pour lesquelles il y a absence totale de synthèse de chaînes adultes et persistance d'une synthèse de chaîne γ fœtale ne permettant pas la compensation de la perte de synthèse de chaîne β, d'où une anémie grave.
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Michel COHEN-SOLAL : maître de recherche à l'I.N.S.E.R.M., C.H.U. Henri-Mondor, Créteil
- Jean-Claude DREYFUS : professeur honoraire à la faculté de médecine Cochin-Port-Royal
Classification
Médias
Hémoglobine : structure quaternaire
Encyclopædia Universalis France
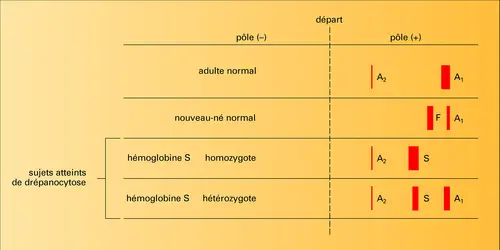
Hémoglobine : électrophorèse
Encyclopædia Universalis France
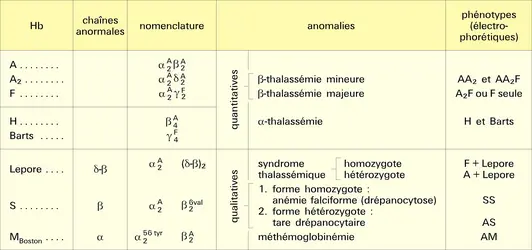
Hémoglobines et hémoglobinoses
Encyclopædia Universalis France
Autres références
-
PATHOLOGIE MOLÉCULAIRE DES HÉMOGLOBINES - (repères chronologiques)
- Écrit par Dominique LABIE
- 189 mots
1949 L. Pauling : isolement par électrophorèse de l'hémoglobine S de la drépanocytose.
1956 V. Ingram : notion de maladie moléculaire (différence de structure entre hémoglobines A et S).
1959 V. Ingram : phylogénie des hémoglobines et démembrement des thalassémies.
1960...
-
ANÉMIES
- Écrit par Bruno VARET
- 3 094 mots
- 5 médias
...– les anémies corpusculaires et constitutionnelles dues à une anomalie de la membrane ( sphérocytose héréditaire, elliptocytose héréditaire...), à une anomalie de l'hémoglobine sans défaut de synthèse (hémoglobine S homozygote, hémoglobine SC, hémoglobine instable) ; à une anomalie enzymatique (déficit... -
CYANOSE
- Écrit par François BOURNÉRIAS
- 145 mots
Coloration bleutée de la peau et des muqueuses, prédominant aux extrémités. Elle résulte d'une augmentation du taux de l'hémoglobine réduite dans le sang périphérique et apparaît lorsque ce taux dépasse 5 grammes pour 100 millilitres.
Il faut distinguer : la cyanose centrale...
-
FIÈVRE BILIEUSE HÉMOGLOBINURIQUE
- Écrit par François BOURNÉRIAS
- 223 mots
-
HÉMATOLOGIE
- Écrit par Jean BERNARD et Michel LEPORRIER
- 8 479 mots
- 1 média
 ...héréditaires, raciales ou familiales, qui atteignent des millions d'hommes en Afrique, en Asie, en Amérique, en Europe méditerranéenne sont dues à la présence d'hémoglobines anormales. Dans la structure de chaque hémoglobine se trouve une chaîne rigoureusement ordonnée d'une centaine d' acides aminés. Le changement...
...héréditaires, raciales ou familiales, qui atteignent des millions d'hommes en Afrique, en Asie, en Amérique, en Europe méditerranéenne sont dues à la présence d'hémoglobines anormales. Dans la structure de chaque hémoglobine se trouve une chaîne rigoureusement ordonnée d'une centaine d' acides aminés. Le changement... - Afficher les 14 références
Voir aussi
- MATURATION DE L'ARN
- HÉRÉDITAIRES MALADIES ou MALADIES GÉNÉTIQUES
- GLOBINE
- HÉMOGLOBINOSES
- TRANSCRIPTION, biologie moléculaire
- TRADUCTION, biologie moléculaire
- DRÉPANOCYTOSE ou ANÉMIE FALCIFORME
- MÉTHÉMOGLOBINE
- POLYPEPTIDIQUE CHAÎNE
- HÈME
- LEPORE HÉMOGLOBINE
- THALASSÉMIE
- HÉMATIE ou GLOBULE ROUGE ou ÉRYTHROCYTE
- CROSSING-OVER, génétique
- PROTÉINES BIOSYNTHÈSE DES
- HOMOZYGOTE
- HÉTÉROZYGOTE
- RÉCESSIVITÉ, génétique
- GÉNÉTIQUE MOLÉCULAIRE
- HÉMOPATHIES
- PROTÉINES
- ÉPISSAGE, génétique moléculaire
