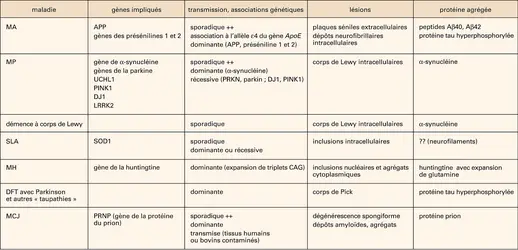
MALADIES NEURODÉGÉNÉRATIVES
Article modifié le
Les maladies neurodégénératives (MND) constituent un groupe de pathologies progressives liées à un dysfonctionnement métabolique au sein du tissu nerveux, conduisant à la mort des neurones et à la destruction du système nerveux. Le cerveau et la moelle épinière peuvent être touchés par des lésions diffuses ou limitées à certaines zones spécifiques. Si certaines de ces maladies atteignent quelquefois l'enfant ou l'adulte jeune, la majorité des cas se rencontrent après 65 ans. Le tableau clinique peut être soit une atteinte prédominante des fonctions psychiques, aboutissant à la démence comme dans la maladie d'Alzheimer ou la maladie de Pick, soit des anomalies motrices prédominantes comme dans la sclérose latérale amyotrophique ou la maladie de Parkinson, soit encore l'association des deux comme dans la chorée de Huntington ou la maladie de Creutzfeldt-Jakob. Pour certaines de ces maladies, des facteurs génétiques ont été mis en cause (formes héréditaires), mais la plupart surviennent de manière isolée (formes sporadiques), ce qui n'exclut pas l'implication de facteurs génétiques. En l'absence de traitement curatif disponible à ce jour, l'évolution se fait vers un état grabataire dont les complications conduisent au décès. Une caractéristique commune à la majorité de ces maladies neurodégénératives est l'accumulation dans le tissu nerveux d'agrégats intra- ou extracellulaires de protéines mal conformées (dépôts amyloïdes), responsables directement ou indirectement de la mort neuronale.
Les avancées dans le diagnostic, la compréhension et la classification des MND ne sont que très récentes. Depuis la première description de la maladie d'Alzheimer (1907) chez une femme présentant une démence et des lésions histologiques particulières (plaques séniles amyloïdes et dépôts fibrillaires dans les neurones), et après l'identification de la maladie de Parkinson en 1912, la nosologie des MND a évolué. La classification fut tout d'abord exclusivement clinique et anatomopathologique : l'association de signes cliniques et de lésions histologiques spécifiques découvertes à l'autopsie définissait chaque maladie. Cette classification, encore utilisée aujourd'hui, fait cependant apparaître des imprécisions et un certain degré de chevauchement entre les différentes pathologies. Depuis une dizaine d'années, des causes génétiques ont été retrouvées dans certaines de ces maladies, dans la maladie d'Alzheimer (MA), la maladie de Parkinson (MP), la chorée de Huntington (CH), la sclérose latérale amyotrophique (SLA), et enfin la maladie à prion ou maladie de Creutzfeldt-Jakob (MCJ), ouvrant ainsi l'ère moderne de la classification « moléculaire » des MND. Depuis lors, on assiste à une explosion de la recherche visant à préciser les mécanismes physiopathologiques impliqués dans la dégénérescence neuronale, ce qui constitue l'étape essentielle à la compréhension et donc au traitement de ces affections.

En 2005, en France, un rapport du Sénat estimait à 855 000 le nombre de personnes âgées de plus de 65 ans présentant une démence, avec une augmentation du nombre de sujets atteints en fonction de l'âge. En raison de l'allongement de la durée de vie, mais aussi des progrès réalisés dans le diagnostic et dans la prise en charge des complications de ces maladies, le nombre de sujets atteints va augmenter dramatiquement dans les années à venir. Les prévisions concernant la France indiquent que ce chiffre sera de 1,3 million en 2020 et de plus de 2 millions en 2040. Pour la maladie d'Alzheimer, la plus fréquente des démences, qui touchait en 2006 plus de 15 millions de personnes dans le monde, ce chiffre devrait tripler d'ici à 2050, avec 13,3 millions de personnes atteintes aux États-Unis et 16,2 millions en Europe. Il est donc évident que les MND vont constituer dans les années à venir un défi médical, social et économique sans précédent.
Les tableaux cliniques
Commençant le plus souvent de façon insidieuse (la date de début est difficile à faire préciser par la famille) chez un sujet auparavant sain, la maladie est fluctuante et progresse lentement avec parfois des périodes de stabilité. Elle sera peu influencée par les traitements actuels, qui sont le plus souvent symptomatiques. Les signes cliniques sont variables selon la maladie, et on distingue trois grands types de tableaux cliniques, que nous allons passer en revue (tableau).
Démence liée à une altération des fonctions cérébrales supérieures
L' exemple type est ici celui de la maladie d'Alzheimer (MA), responsable de plus de la moitié des cas de démence de la personne âgée (cf. maladie d'alzheimer). La prévalence de la MA est de 6,1 p. 100 chez les hommes et 8,9 p. 100 chez les femmes de plus de 65 ans. Ces chiffres s'élèvent respectivement à 13,2 p. 100 et 20,5 p. 100, après 75 ans, et à près de 30 p. 100 après 80 ans. La maladie débute par une altération progressive de la mémoire, d'apparition insidieuse, car souvent méconnue ou banalisée, rapportée au vieillissement du sujet. Les troubles de la mémoire portent d'abord sur les faits récents, la mémoire des faits anciens étant initialement bien conservée. Il s'y associe progressivement d'autres anomalies : un manque du mot, des difficultés d'orientation temporo-spatiale, des troubles du langage, une apraxie (trouble de l'exécution des gestes, malgré une compréhension et des fonctions motrices intactes), une agnosie (impossibilité d'identifier un stimulus sonore ou visuel) et enfin des troubles comportementaux, aboutissant à une perte progressive de l'autonomie.
Histologiquement, outre une atrophie diffuse du cerveau, deux types de lésions (fig. 1) coexistent :
– des dépôts extracellulaires, les plaques séniles (plaques amyloïdes), constitués de fragments protéiques (peptide Aβ), provenant du clivage anormal d'une glycoprotéine des membranes cellulaires appelée APP (amyloid precursor protein) ;
– des dépôts fibrillaires intra-neuronaux constitués, entre autres, d'une forme hyperphosphorylée de la protéine tau, protéine impliquée dans le transport axonal ; il en résulte une dégénérescence neurofibrillaire (fig. 2).
La maladie est le plus souvent sporadique, mais de rares formes familiales ont été décrites, liées à des mutations du gène de l'APP ou d'autres gènes impliqués dans son métabolisme (préséniline 1 et 2).
La dégénérescence fronto-temporale (DFT), qui est dans nombre de cas la deuxième cause de démence dégénérative après la MA, correspond à plusieurs entités histologiques ayant en commun une atrophie cérébrale prédominant sur les lobes frontaux et pariétaux. Cliniquement, les DFT sont caractérisées par des troubles comportementaux relativement précoces (entre 50 et 60 ans). La plus classique des DFT (mais qui n'est pas la plus fréquente) est la maladie de Pick, caractérisée par la présence de corps de Pick (agrégats de protéine tau). Dans certains cas familiaux, les troubles comportementaux sont associés à un syndrome parkinsonien. Des dépôts fibrillaires d'une forme mutée de la protéine tau y ont été retrouvés.
Démence où l'atteinte motrice est au premier plan
Il peut s'agir d'une perte progressive des fonctions motrices, ou de l'apparition de mouvements anormaux.
La maladie de Parkinson est la deuxième cause de MND ; elle est due à la dégénérescence d'une population de neurones qui sont situés dans la substance noire mésencéphalique et qui produisent un neurotransmetteur, la dopamine. Elle touche plus de 2 p. 100 des sujets de plus de 65 ans, et on dénombre en France plus de 10 000 nouveaux cas par an. Le tableau clinique caractéristique associe un tremblement de repos, une hypertonie (rigidité), une akinésie (difficulté à initier ou à exécuter des mouvements) aboutissant à des troubles de la marche et de la posture. Histologiquement, la lésion caractéristique est la présence de corps de Lewy dans les neurones, constitués d'agrégats d'une protéine, l'α-synucléine. La maladie est le plus souvent sporadique ; de rares formes familiales ont cependant été décrites, causées par des mutations dans le gène de l'α-synucléine.
La sclérose latérale amyotrophique (SLA) est due à une atteinte dégénérative des neurones moteurs, entraînant une perte progressive des fonctions motrices. Les premiers signes cliniques apparaissent en général entre 50 et 60 ans, avec une prévalence de 4 à 6 pour 100 000 habitants. Le tableau clinique associe une amyotrophie (fonte musculaire), un déficit moteur (débutant le plus souvent au niveau des extrémités des membres supérieurs pour s'étendre secondairement à l'ensemble de la musculature), des crampes, des fasciculations (contractions brèves des faisceaux musculaires), une atteinte des nerfs crâniens. L'association d'une rigidité (spasticité) des membres avec des réflexes ostéo-tendineux vifs (syndrome pyramidal) témoigne de l'atteinte des nerfs moteurs.
Démence progressive associée à des anomalies neurologiques
La chorée de Huntington (CH), ou danse de Saint-Guy, qui touche moins de 1 personne sur 5 000, est une maladie héréditaire caractérisée par l'association d'une démence progressive et de mouvements anormaux choréiques (mouvements brusques et imprévisibles, souvent de rotation) et athétosiques (mouvements lents de torsion et d'ondulation des membres). Les premiers signes apparaissent entre 30 et 50 ans. La CH est liée à une anomalie génétique caractérisée par la répétition anormale d'une partie de l'information génétique (triplets CAG) contenue dans le gène d'une protéine, la huntingtine, et fait partie des maladies dites à « expansion de triplets » (voir plus loin). Des agrégats de huntingtine anormale sont retrouvés dans les noyaux gris centraux du cerveau, structures qui exercent une action facilitatrice sur les mouvements en focalisant les informations provenant du cortex, mais aussi une action inhibitrice en bloquant la réalisation des mouvements lorsque ceux-ci sont inadaptés.
La démence à corps de Lewy est une maladie intermédiaire entre la maladie d'Alzheimer et la maladie de Parkinson, conjuguant les signes cliniques de ces deux pathologies. Le début est un peu plus précoce que dans la MA, l'évolution peut être plus rapide. La lésion caractéristique est la présence d'inclusions neuronales, les corps de Lewy, similaires à ceux qui sont retrouvés dans la MP.
La maladie de Creutzfeldt-Jakob (MCJ) ou encéphalopathie spongiforme subaiguë (ESS) est, quant à elle, une maladie rare, due à l'accumulation dans le cerveau d'une forme anormale de la protéine prion, glycoprotéine qui existe normalement dans le cerveau de l'homme. La maladie touche l'adulte de 50 à 75 ans, et débute par des troubles psychiatriques évoluant rapidement vers la démence. Il s'y associe divers signes neurologiques. L'évolution vers l'état grabataire et le décès est rapide. La MCJ est dans la majorité des cas (85 p. 100) sporadique, une origine génétique étant retrouvée dans 10 à 15 p. 100 des cas (formes familiales). Dans moins de 5 p. 100 des cas, elle est transmise par consommation de viande de bovins atteints par l'encéphalopathie spongiforme bovine (ESB) ou par inoculation de produits humains contaminés par des protéines prions ; elle peut alors toucher des patients beaucoup plus jeunes (cf. encéphalopathies spongiformes).
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Nathalie CARTIER-LACAVE : directeur de recherche à l'I.N.S.E.R.M., pédiatre, consultant à l'hôpital Saint-Vincent-de-Paul, Paris
- Caroline SEVIN : auteur
Classification
Médias
Maladies neurodégénératives
Encyclopædia Universalis France
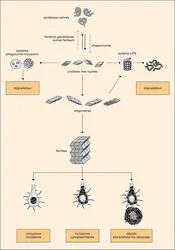
Mécanisme d'accumulation des protéines anormales dans le tissu nerveux
Encyclopædia Universalis France
Autres références
-
ALZHEIMER MALADIE D'
- Écrit par Nathalie CARTIER-LACAVE
- 1 872 mots
C' est le psychiatre allemand Emil Kraepelin, en 1912, dans son Traité de psychiatrie, qui a donné le nom de maladie d'Alzheimer à cette démence dégénérative affectant le sujet « jeune ». Cet état pathologique a été décrit en effet pour la première fois en 1907 par son confrère et compatriote...
-
AMNÉSIE
- Écrit par Francis EUSTACHE
- 1 108 mots
Les amnésies constituent un terme générique qui s’applique à de multiples situations pathologiques : une maladie neurodégénérative, comme la maladie d’Alzheimer, un traumatisme crânien, les conséquences de lésions focales de diverses origines, comme une pathologie infectieuse, vasculaire, tumorale,...
-
CERVEAU ET BILINGUISME
- Écrit par Jean-Marie ANNONI
- 804 mots
- 1 média
 ...structures semblent davantage développées chez le bilingue actif (qu’il soit précoce ou tardif) et améliorent sa réserve cognitive, ce qui expliquerait pourquoi les bilingues résisteraient mieux aux maladies neurodégénératives comme la maladie d’Alzheimer, en tout cas dans les premières phases.
...structures semblent davantage développées chez le bilingue actif (qu’il soit précoce ou tardif) et améliorent sa réserve cognitive, ce qui expliquerait pourquoi les bilingues résisteraient mieux aux maladies neurodégénératives comme la maladie d’Alzheimer, en tout cas dans les premières phases. -
CYANOBACTÉRIES ou CYANOPHYCÉES, anc. ALGUES BLEUES
- Écrit par Pierre BOURRELLY et Jean Claude LEFEUVRE
- 2 711 mots
- 3 médias
 ...Consommées, ces roussettes seraient en partie responsables (les Chamorros utilisant également de la farine préparée à partir de ces mêmes graines de Cycas), chez certains patients, d'une sclérose latérale amyotrophique, associée à un complexe de symptômes de type maladie de Parkinson et de signes...
...Consommées, ces roussettes seraient en partie responsables (les Chamorros utilisant également de la farine préparée à partir de ces mêmes graines de Cycas), chez certains patients, d'une sclérose latérale amyotrophique, associée à un complexe de symptômes de type maladie de Parkinson et de signes... - Afficher les 20 références
Voir aussi
- MÉMOIRE TROUBLES DE LA
- MUTATIONS DYNAMIQUES ou MALADIES À EXPANSION DE TRIPLETS
- NEUROLOGIE CLINIQUE
- HÉRÉDITAIRES MALADIES ou MALADIES GÉNÉTIQUES
- APOPTOSE
- PRION
- VIEILLESSE
- PICK MALADIE DE
- CREUTZFELDT-JAKOB MALADIE DE
- MALADIES MITOCHONDRIALES
- THÉRAPIE CELLULAIRE
- AKINÉSIE
- DÉGÉNÉRATIVES MALADIES
- AGNOSIE
- HYPERTONIE MUSCULAIRE
- HISTOLOGIE PATHOLOGIQUE
- CELLULES SOUCHES EMBRYONNAIRES
- SCLÉROSE LATÉRALE AMYOTROPHIQUE
- UBIQUITINE
- ANATOMIE PATHOLOGIQUE
- CELLULES SOUCHES ADULTES
- THÉRAPIE GÉNIQUE
- NEURONE ou CELLULE NERVEUSE
- DÉGÉNÉRESCENCE FRONTO-TEMPORALE
- LEWY DÉMENCE À CORPS DE
- PROTÉINES REPLIEMENT DES
- PEROXYSOME
- L-DOPA
- CHAPERONNES PROTÉINES
- ENZYMOPATHIES
- MOTONEURONE ou NEURONE MOTEUR
- MOLÉCULES BIOLOGIQUES, structure et fonction
- TREMBLEMENT
- PROTÉINES
- DÉFICITS ENZYMATIQUE & MÉTABOLIQUE
- PROTÉINE PRION (PrP)
- PARKINSONIEN SYNDROME
- ALPHA-SYNUCLÉINE
