MATIÈRE (physique) État gazeux
Article modifié le
Les gaz réels en équilibre thermodynamique
Forces intermoléculaires
La nature des forces intermoléculaires est d'origine électromagnétique et résulte de l'existence de charges électroniques et nucléaires. L'interaction entre deux molécules séparées par la distance r s'exprime par l'énergie potentielle V(r) ou par la force F(r) reliée au potentiel par :
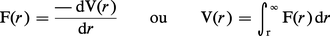

Énergie d'interaction de deux molécules
Encyclopædia Universalis France
En général, V(r) et F(r) dépendent également de l'orientation relative des molécules. Le potentiel intermoléculaire V(r) possède la forme générale représentée sur la figure 3. Cette forme traduit l'existence de forces attractives à longue portée (r ~ 1 nm) et répulsives à courte portée (r ~ 0,1 nm). Les premières, appelées forces de Van der Waals, sont responsables de la condensation des gaz lorsque leur température est suffisamment basse. Les secondes sont à l'origine de la compressibilité limitée de la matière condensée. L'idée que les molécules se repoussent à courte distance et s'attirent à longue distance a été proposée pour la première fois par le physicien allemand Rudolf Clausius en 1857. Mais ce n'est que grâce à la mécanique quantique qu'on a pu arriver, vers 1930, à une compréhension satisfaisante de la nature des forces intermoléculaires. Leur description précise, entreprise depuis le début des années 1960, est l'un des objectifs majeurs de la physique moléculaire.
Forces attractives
Un atome, ou une molécule, constitue une distribution de charges caractérisée par ses moments multipolaires :

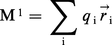
L'énergie potentielle d'interaction s'exprime par une série de termes faisant intervenir les moments d'ordres croissants et l'orientation relative des molécules. Le premier terme non nul décrivant l'interaction entre deux molécules neutres fait intervenir leurs moments dipolaires MA1 et MB1. Dans un gaz, la moyenne de l'énergie d'interaction dipôle-dipôle effectuée sur toutes les orientations possibles vaut :
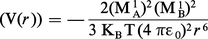

Les termes suivants, décrivant les interactions dipôle-quadrupôle, quadrupôle-quadrupôle..., sont en général plus faibles. Les forces attractives entre multipôles permanents constituent les forces d'orientation.
En présence d'un champ électrique E, la distribution de charge d'une molécule est modifiée. Il apparaît un moment dipolaire induit μ = αE, où α est la polarisabilité de la molécule dont l'ordre de grandeur est 4πε0 fois son volume, soit 10—40 F.m2. Il en résulte une interaction dipôle-dipôle induit dont l'énergie moyenne a pour expression :
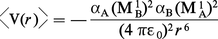
Une molécule dépourvue de moments électriques permanents possède néanmoins un moment dipolaire instantané résultant du mouvement des électrons par rapport aux noyaux. Bien que la valeur de ce moment soit nulle en moyenne, la valeur moyenne du produit des moments de deux molécules voisines ne l'est pas. L'énergie d'interaction qui résulte de cet effet purement quantique vaut en première approximation :

Les forces de Van der Waals sont très faibles par rapport aux forces intramoléculaires. Dans une molécule stable, l'énergie de liaison est de l'ordre de quelques électronsvolts (de 4 à 12 eV). Pour deux molécules identiques possédant un moment dipolaire μ = 1 D et à T = 300 K, la valeur moyenne de l'énergie d'interaction dipôle-dipôle vaut 6,5 × 10—4 eV à la distance r = 0,5 nm et 1,0 × 10—5 eV à r = 1 nm. La polarisabilité des petites molécules (α/4πε0) est de l'ordre de 3 × 10—3 (nm)3. Avec cette valeur, l'énergie moyenne d'interaction dipôle-dipôle induit est de 1,2 × 10—4 eV à 0,5 nm et 1,9 × 10—6 eV à 1 nm. Enfin, le terme prépondérant de l'énergie de dispersion en — C/R6, avec C ≈ 5 × 10—78 Jm—6 pour des molécules usuelles telles que Ar, O2, N2, vaut 2 × 10—3 eV à 0,5 nm et 3 × 10—5 eV à 1 nm. À l'exception des molécules fortement polaires comme H2O (μ = 1,85 D), la contribution dominante provient des forces de dispersion. L'énergie d'induction est généralement faible.
Forces répulsives
Lorsque deux molécules sont suffisamment proches pour que leurs nuages électroniques se recouvrent, le principe d'exclusion de Pauli, qui interdit à deux électrons occupant le même état quantique de se trouver dans la même région de l'espace, entraîne une diminution de la densité électronique dans la région de recouvrement. La charge positive des noyaux atomiques n'est plus totalement écrantée par la charge négative électronique et ceux-ci se repoussent. L'énergie de répulsion n'est pas facile à calculer. La contribution principale est une quantité appelée intégrale d'échange.
À très courte distance, l'énergie d'interaction varie en 1/r ; à plus grande distance, elle est proportionnelle à

Représentation des forces intermoléculaires
La relation entre le potentiel intermoléculaire et les grandeurs macroscopiques observables est très complexe. De plus, celles-ci sont peu sensibles à la forme exacte du potentiel. Aussi, pour le décrire, on utilise une fonction analytique simple contenant un petit nombre de paramètres que l'on ajuste à partir des données expérimentales. De nombreuses formes empiriques de potentiel ont été proposées. La plus simple d'entre elles décrit les collisions élastiques entre des sphères rigides de diamètre d : V(r) = ∞ pour (r < d) et V(r) = 0 pour (r ≥ d).
Ce potentiel, exclusivement répulsif, a été abondamment utilisé pour établir les principes de la théorie cinétique. La manière la plus simple de prendre en compte les forces attractives est de rajouter au potentiel précédent un puits de profondeur constante, c'est-à-dire : V(r) = ∞ (r ≤ d1), V(r) = — ε (d1 < r < d2) et V(r) = 0 (r ≥ d2).
Un potentiel plus réaliste est celui qui a été proposé par le physicien britannique John Edward Lennard-Jones (1930) :

Le terme attractif en r—6 décrit correctement les forces d'induction et de dispersion. Le choix du terme répulsif en r—12, qui diffère notablement de la forme exponentielle suggérée par la théorie, est plus arbitraire. Ce potentiel, appelé LJ(12-6), possède néanmoins l'avantage considérable d'être d'une grande simplicité. Il est d'une extrême utilité pour l'interprétation théorique des propriétés de l'état gazeux. De nombreuses autres formes de potentiel ont été proposées, avec par exemple substitution d'un terme Ae—Br à celui en r—12 et augmentation jusqu'à 9 du nombre de paramètres ajustables. Malgré ces améliorations, il n'est pas possible d'obtenir une forme analytique convenable pour une valeur quelconque de la distance intermoléculaire.
À partir de 1970, des progrès considérables ont été réalisés dans la connaissance des potentiels intermoléculaires. Grâce à l'augmentation spectaculaire de la vitesse de calcul des ordinateurs, on est maintenant capable de déterminer numériquement des surfaces d'énergie potentielle qui décrivent correctement l'anisotropie de l'interaction entre molécules non sphériques. Les travaux combinent désormais les méthodes de la mécanique quantique (calculs ab initio) avec les techniques classiques (calculs de dynamique moléculaire). L'amélioration de la précision des mesures et l'apparition de nouvelles techniques expérimentales permettent de tester les potentiels théoriques avec une grande finesse. De nombreuses propriétés des gaz sont utilisées simultanément : leurs écarts à la loi des gaz parfaits, leurs propriétés de transport, celles des gaz condensés, la diffusion d'un jet moléculaire monocinétique par un gaz ou par un second jet croisant le premier à angle droit, les spectres de molécules de Van der Waals, etc.
Équation d'état des gaz réels
Les premières expériences qui ont montré qu'aucun gaz réel ne suit exactement la loi de Boyle-Mariotte ont été réalisées en 1847 par Victor Regnault (1810-1878). L'étude de la compressibilité des gaz a été ensuite poursuivie pendant plus d'un siècle, avec une précision croissante qui atteint 10—4 dans les expériences modernes. Les déviations des gaz réels par rapport au gaz parfait sont mises en évidence sur un diagramme d'Amagat, où l'on porte le facteur de compressibilité Z = PVm/RT en fonction de la pression P et où Vm est le volume molaire. Aux faibles pressions, tous les gaz satisfont à la loi des gaz parfaits (Z ≈ 1) avec une bonne précision (mieux que 1 p. 100 pour des gaz simples tels que le diazote N2 ou l'argon Ar à température ordinaire et pour des pressions inférieures à 15 atm). À haute température, les gaz réels sont moins compressibles que le gaz parfait (Z > 1). À pression plus élevée, tous les gaz, excepté l'hydrogène, sont plus compressibles que ne le prédit la loi de Mariotte (Z < 1). À température suffisamment basse, les isothermes présentent un minimum qui traduit la diminution de compressibilité lorsqu'on s'approche de l'état liquide. Les propriétés de nombreux gaz réels peuvent être représentées approximativement par une équation d'état universelle f(P, V, T) = 0 fonction des variables réduites P = P/Pc, V = V/Vc et T = T/Tc, où Pc, Vc et Tc sont les valeurs critiques de la pression, du volume et de la température (principe des états correspondants).
De nombreuses équations d'état ont été proposées pour décrire les gaz réels. La plupart d'entre elles reposent sur deux idées suggérées par la théorie cinétique et l'existence des forces intermoléculaires. La première est que seul l'espace intermoléculaire vide de matière est susceptible de varier en fonction de la température ou de la pression. Une telle hypothèse revient à prendre en compte la répulsion des molécules lorsqu'elles sont en contact. Si l'on considère les molécules comme des sphères rigides, le volume inaccessible au centre de deux molécules identiques de diamètre d, lors d'une collision binaire, vaut (4/3)πd3, soit (2/3)πd3 par molécule. La réduction du volume molaire qui en résulte porte le nom de covolume et vaut : b = 2/3(πNAd3).
La seconde idée consiste à prendre en compte la contribution de l'attraction mutuelle des molécules à la pression interne du gaz qui vient s'ajouter à celle des collisions moléculaires sur les parois du récipient. L'augmentation de pression correspondante est proportionnelle au nombre d'interactions binaires, c'est-à-dire au carré de la densité moléculaire, soit ΔP = a/Vm2, a étant une constante. On obtient ainsi l'équation de Van der Waals (1877) :
Une autre équation d'état, appelée équation du viriel, exprime le facteur de compressibilité sous la forme d'un développement en série de la densité moléculaire :





Le coefficient B(T) est négatif à basse température et positif à haute température. Il s'annule pour une valeur de T appelée température de Boyle. Le gaz réel se comporte alors approximativement comme un gaz parfait.
Accédez à l'intégralité de nos articles
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Henri DUBOST : docteur ès sciences, directeur de recherche au C.N.R.S.
- Jean-Marie FLAUD : docteur ès sciences, directeur de recherche au C.N.R.S.
Classification
Médias

États désordonnés de la matière
Encyclopædia Universalis France

Représentation du diagramme de phase d'un corps pur
Encyclopædia Universalis France

Température d'ébullition des gaz rares
Encyclopædia Universalis France
Autres références
-
ÉTAT DE LA MATIÈRE, notion d'
- Écrit par Bernard PIRE
- 1 521 mots
L'expérience quotidienne permet à chacun d'appréhender la notion d'état de la matière (parfois appelé phase) et celle de transition de phase qui lui est étroitement liée. L'exemple typique est celui des trois états si différents que prend l'eau lorsque sa température varie : à partir de 0 ...
-
MATIÈRE, notion de
- Écrit par Jean-Marc LÉVY-LEBLOND
- 2 022 mots
Le mot « matière » cache sous sa généralité abstraite une origine concrète fort éclairante. En latin archaïque, materia appartient à la langue rustique et désigne la substance dont est fait le tronc de l'arbre, en tant qu'elle est productrice (de branches, de feuilles). L'élargissement successif...
-
ANTIMATIÈRE
- Écrit par Bernard PIRE et Jean-Marc RICHARD
- 6 934 mots
- 4 médias
 ...associé est bâti avec les antiquarks correspondants. Les mésons résultent de la liaison d'un quark et d'un antiquark. Dans cette description moderne, la matière est constituée par trois « générations » de quarks et de leptons, le nombre trois a été établi par les expériences du C.E.R.N. (laboratoire européen...
...associé est bâti avec les antiquarks correspondants. Les mésons résultent de la liaison d'un quark et d'un antiquark. Dans cette description moderne, la matière est constituée par trois « générations » de quarks et de leptons, le nombre trois a été établi par les expériences du C.E.R.N. (laboratoire européen... -
ATOME
- Écrit par José LEITE LOPES
- 9 146 mots
- 13 médias
L'atome est le terme ultime de la division de la matière dans lequel les éléments chimiques conservent leur individualité. C'est la plus petite particule d'un élément qui existe à l'état libre ou combiné. On connaît 90 éléments naturels auxquels s'ajoutent le ...
-
BOHR ATOME DE
- Écrit par Bernard PIRE
- 369 mots
- 1 média

Deux ans après avoir soutenu sa thèse sur la théorie électronique des métaux, le physicien danois Niels Bohr (1885-1962) écrit en 1913 trois articles fondamentaux qui révolutionnent la compréhension de la structure de la matière. Le premier, paru le 5 avril dans le Philosophical Magazine...
-
ATOME, notion d'
- Écrit par Bernard PIRE
- 1 499 mots
« Brique insécable » de la matière selon son étymologie, l'atome est le terme ultime de la division de la matière dans lequel les éléments chimiques conservent leur individualité. C'est la plus petite particule d'un élément qui existe à l'état libre ou combiné, les corps simples étant...
- Afficher les 40 références
Voir aussi
- ONDE ou RAYONNEMENT ÉLECTROMAGNÉTIQUE
- GAZ DIATOMIQUES
- GAZ POLYATOMIQUES
- ABSORPTION, physique
- PRESSION, physique
- POLARISABILITÉ MOLÉCULAIRE
- UNITÉS SYSTÈMES D'
- ÉTATS DE LA MATIÈRE
- LIBRE PARCOURS MOYEN
- POTENTIEL INTERMOLÉCULAIRE
- INTERACTIONS MOLÉCULAIRES
- ISOBARE
- PHASE TRANSITIONS DE
- VITESSE
- ÉTAT CHANGEMENT D'
- COLLISION, physique
- COLLISIONS ÉLASTIQUES
- ÉQUILIBRE THERMODYNAMIQUE
- MOLÉCULES DIATOMIQUES
- DIFFUSION
- ÉTAT ÉQUATION D'
- ÉNERGIE INTERNE
- DIAGRAMME, thermodynamique
- POINT CRITIQUE
- EXCITATION ÉLECTRONIQUE ÉNERGIE D'
- CHARLES LOI DE
- COMPRESSIBILITÉ ISOTHERME COEFFICIENT DE
- CLAPEYRON DIAGRAMME DE
- FUSION
- MAXWELL-BOLTZMANN DISTRIBUTION DE
- GAZ MONOATOMIQUES
- GAZ RÉELS
- GAZ PARFAITS LOI DES
- LOSCHMIDT NOMBRE DE
- GAY-LUSSAC LOI DE
- ISOTHERME
- VAN DER WAALS ÉQUATION DE
- POLAIRES MOLÉCULES
- TRANSMISSION, optique
- VAPORISATION
- SUBLIMATION, thermodynamique
- POINT TRIPLE, thermodynamique
- MOLÉCULES POLYATOMIQUES
- THERMIQUE AGITATION
- ROTATION MOLÉCULAIRE ÉNERGIE DE
- VIBRATION MOLÉCULAIRE ÉNERGIE DE
- VARIABLES D'ÉTAT
- BEER-LAMBERT LOI DE
- TRANSITION, physique
- ÉNERGIE POTENTIELLE
- PASCAL, unité
- ÉPAISSEUR OPTIQUE
- ENTHALPIE
- GAZEUX ÉTAT
- FONCTION D'ÉTAT, thermodynamique
- TRAVAIL, physique
- TEMPÉRATURE
- BOLTZMANN CONSTANTE DE
- PARTITION FONCTION DE
- ÉNERGIE LIBRE DE HELMHOLTZ
- NIVEAU, physique atomique
- ISOCHORE
- JOULE LOIS DE, thermodynamique
- BOYLE-MARIOTTE LOI DE
- ADIABATIQUE TRANSFORMATION
- CAPACITÉ CALORIFIQUE
- AMAGAT DIAGRAMME D'
- LONDON FORCES DE
- VIRIEL ÉQUATION DU
- ÉNERGIE CINÉTIQUE
- DISTRIBUTION DE CHARGE
- PHASES, physico-chimie
- TEMPÉRATURE CRITIQUE
- TRANSPORT ÉQUATION DE
- TRANSFORMATIONS THERMODYNAMIQUES
- AVOGADRO NOMBRE D'
- RAIE SPECTRALE
- SPECTRE D'ABSORPTION
- MOMENT ÉLECTRIQUE
- COEFFICIENT DE DIFFUSION
- CONDUCTION THERMIQUE
- TORR ou MILLIMÈTRE DE MERCURE, unité
