
- 1. Présentation clinique de la mucoviscidose
- 2. Épidémiologie de la mucoviscidose
- 3. Le gène et la protéine CFTR
- 4. Les relations génotype CFTR/phénotype clinique
- 5. Histoire et géographie des mutations du gène CFTR
- 6. Diagnostic précoce de la mucoviscidose
- 7. Les traitements de la mucoviscidose : la révolution des thérapies « mutations spécifiques »
- 8. Bibliographie
- 9. Sites internet
MUCOVISCIDOSE ou FIBROSE KYSTIQUE DU PANCRÉAS
Le gène et la protéine CFTR
Les années 1990 ont été marquantes dans la découverte des gènes de maladies génétiques. Identifié en 1989, le gène de la mucoviscidose a été, avec celui de l’hémophilie et celui de la myopathie de Duchenne, parmi les tout premiers gènes de maladie monogénique à avoir été clonés.
Pour identifier le gène responsable de la mucoviscidose, une stratégie dite « de génétique inverse » (ou encore de clonage positionnel) a été mise en œuvre. La protéine mutée dans cette maladie étant inconnue, il a fallu tout d’abord localiser le gène sur l’un des 22 chromosomes autosomes, puis s’en rapprocher en étudiant la liaison du gène CF avec des gènes ou des marqueurs de localisation chromosomique connue pour, in fine, l’identifier. En 1985, une première étape est franchie par une équipe canadienne de l’Hospital for Sick Children à Toronto, dirigée par Lap-Chee Tsui, qui localise, grâce à une analyse de liaison génétique, le gène CF sur le bras long du chromosome 7, en 7q35, région chromosomique qui contient par ailleurs plusieurs gènes liés à la physiologie gastro-intestinale.
Un consortium de laboratoires européens dirigé par Robert Williamson, à Londres, entre alors dans la compétition et est près d’aboutir, mais, malgré malgré l’excellent travail de celui-ci, c’est finalement, en 1989, l’équipe américano-canadienne coordonnée par Tsui, Riordan et Collins qui réussit le tour de force d’isoler (de cloner) le gène CF responsable – qui devient CFTR pour cysticfibrosistransmembrane conductance regulator, du fait des caractéristiques fonctionnelles de la protéine pour laquelle il code. Ces résultats sont publiés dans trois articles successifs de Science, ouvrant de nouvelles perspectives pour comprendre la maladie et imaginer des stratégies thérapeutiques.
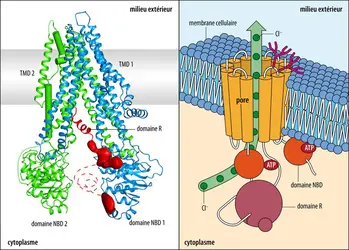
Structure tridimensionnelle et fonctionnement de la protéine CFTR
à gauche : Laboratory of Membrane Biology and Biophysics at The Rockefeller University ; à droite :EUF
CFTR est un gène d’assez grande taille, long d’environ 180 000 nucléotides. Sa partie codante est organisée en 27 exons qui définissent une protéine transmembranaire de 1 480 acides aminés, appartenant à la famille des transporteurs membranaires ABC, c’est-à-dire des protéines qui utilisent l’adénosine triphosphate (ATP) comme source d’énergie pour réaliser un transport actif d’ions et de molécules organiques au travers de la membrane cellulaire. La protéine CFTR est un canal chlorure qui régule le passage vers l’espace extracellulaire des ions chlorure et bicarbonate au niveau des cellules épithéliales de l’organisme, en particulier au niveau pulmonaire et pancréatique : les ions Cl- qui pénètrent dans la cellule par la membrane basale en même temps que les ions Na+ et K+ sont expulsés au travers de la partie apicale de la cellule par la protéine CFTR. Cette dernière est engagée dans un complexe de protéines membranaires, et son activité (c’est-à-dire l’ouverture plus ou moins marquée du canal) est régulée entre autres par l’adénosine monophosphate (AMP) cyclique. La protéine présente deux sites de liaison à l’ATP et un domaine de régulation de son activité, le domaine R.
Une mutation fréquente, la perte de trois nucléotides correspondant au codon 508 de la protéine (Δp.Phe508 = c.1521 – 1523del CTT) situé dans l’exon 10, est présente chez 70 % des allèles mutés en France et est décrite dans la publication princeps de Science. Elle se traduit par la perte d’un seul acide aminé (la phénylalanine), ce qui empêche la protéine de se replier de manière correcte ; elle est alors retenue sur son lieu de synthèse dans le réticulum endoplasmique et dégradée.
Immédiatement après la découverte du gène et de sa principale mutation, Lap-Chee Tsui propose la création d’un consortium international d’études des mutations (Cystic Fibrosis Genetic Analysis Consortium). Un peu plus tard, une seconde base de données associant des données fonctionnelles aux variants identifiés est[...]
La suite de cet article est accessible aux abonnés
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Claude FÉREC : professeur des Universités, praticien hospitalier (PU/PH), professeur émérite de génétique médicale, université de Brest
Classification
Médias

Malade atteint de mucoviscidose
RyanJLane/ getty Images
Atteintes pulmonaires et pancréatiques de la mucoviscidose
Claude Ferec/ INSERM
Structure tridimensionnelle et fonctionnement de la protéine CFTR
à gauche : Laboratory of Membrane Biology and Biophysics at The Rockefeller University ; à droite :EUF
Autres références
-
ARNm THÉRAPEUTIQUES
- Écrit par Bruno PITARD
- 6 616 mots
- 5 médias
 Les thérapies de remplacement sont en essai clinique pour le traitement de la mucoviscidose. Les patients atteints de mucoviscidose souffrent d’infections pulmonaires répétées et de problèmes respiratoires chroniques dus au défaut de la protéine CFTR (cysticfibrosistransmembrane conductance...
Les thérapies de remplacement sont en essai clinique pour le traitement de la mucoviscidose. Les patients atteints de mucoviscidose souffrent d’infections pulmonaires répétées et de problèmes respiratoires chroniques dus au défaut de la protéine CFTR (cysticfibrosistransmembrane conductance... -
HÉRÉDITÉ
- Écrit par Charles BABINET , Luisa DANDOLO , Jean GAYON et Simone GILGENKRANTZ
- 11 231 mots
- 7 médias
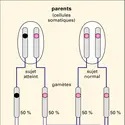 ...risque sur 4 d'avoir, à chaque nouvelle naissance, un enfant atteint. Les enfants indemnes de la fratrie ont une probabilité de 2/3 d'être hétérozygotes. Un bon exemple en est la mucoviscidose (ou fibrose kystique du pancréas), maladie récessive la plus fréquente dans les populations européennes. Cette maladie...
...risque sur 4 d'avoir, à chaque nouvelle naissance, un enfant atteint. Les enfants indemnes de la fratrie ont une probabilité de 2/3 d'être hétérozygotes. Un bon exemple en est la mucoviscidose (ou fibrose kystique du pancréas), maladie récessive la plus fréquente dans les populations européennes. Cette maladie... -
MALADIES MOLÉCULAIRES
- Écrit par Jean-Claude DREYFUS et Fanny SCHAPIRA
- 6 786 mots
- 1 média
Un modèle de maladie autosomique récessive, la mucoviscidose (ou fibrose kystique). C'est la plus fréquente des maladies récessives en Europe, touchant un enfant pour 2 500 naissances, ce qui signifie que la fréquence des porteurs sains hétérozygotes est de 1 sur 25. Elle entraîne deux symptômes... -
MYCOBACTÉRIES
- Écrit par Carlo COCITO , Encyclopædia Universalis et Gabriel GACHELIN
- 4 331 mots
- 5 médias
 ...mycobactérioses aggravant des affections pulmonaires chroniques (silicose par ex.) est sérieux. Mais il est encore plus à craindre pour les patients atteints de mucoviscidose : Mycobacterium avium est ici le plus souvent en cause. Le traitement se fonde sur la sensibilité des souches à la clarithromycine, et...
...mycobactérioses aggravant des affections pulmonaires chroniques (silicose par ex.) est sérieux. Mais il est encore plus à craindre pour les patients atteints de mucoviscidose : Mycobacterium avium est ici le plus souvent en cause. Le traitement se fonde sur la sensibilité des souches à la clarithromycine, et... - Afficher les 7 références
