
- 1. L’ADN, molécule support de l’information génétique
- 2. Le séquençage « historique et classique » de l’ADN : la méthode de Sanger
- 3. Le séquençage NGS et ses approches à haut débit
- 4. La troisième génération de séquençage
- 5. Le single cell sequencing (SCS)
- 6. Ordonner les données de séquençage
- 7. Exploiter les données de NGS
- 8. Évolution des techniques et perspectives
- 9. Bibliographie
SÉQUENÇAGE HAUT DÉBIT DE L'ADN
Article modifié le
Ordonner les données de séquençage

Alignement des séquences d’ADN déterminées
Encyclopædia Universalis France
Le NGS requiert systématiquement un traitement bio-informatique pour gérer le type, la qualité et la quantité de données générées – les capacités de stockage et de traitement de l’information doivent être adéquates ! – afin de pouvoir les interpréter. Les fichiers bruts contenant des milliers voire des millions de séquences sont filtrés afin d’éliminer les données de mauvaise qualité. Le plus souvent, les séquences restantes (reads ou « lectures ») sont ensuite alignées par rapport à un génome ou un transcriptome de référence annoté et présent dans les bases de données (mapping). Si ce n’est pas le cas, ce qui est de plus en plus rare, elles peuvent être alignées seulement entre elles afin de reconstruire la séquence de l’ADN (ou de l’ARN) initiale que l’on souhaite étudier en exploitant leurs zones de chevauchement. Ces alignements peuvent être facilités si le séquençage a été fait dans les deux sens (paired end) sur les fragments de la librairie, et d’autant plus si la librairie a été construite avec une taille calibrée supérieure à 2 kb (technologie mate pair).
Il est préférable que chaque base de la séquence de départ à analyser ait été lue de nombreuses fois pour avoir une certaine profondeur de séquençage (exprimée en X). Ainsi, une base lue 30 fois (à travers 30 lectures) aura une profondeur de 30X et donc une certaine fiabilité, surtout si les lectures ont été faites dans les deux sens. En fonction des applications, une profondeur moyenne minimale sera requise, par exemple 25X pour du séquençage de novo, 10-15X pour effectuer la recherche de SNP – single nucleotidepolymorphism, variation d’une seule base au niveau des chromosomes homologues d’un individu ou entre individus de la même espèce. De même, la totalité de la séquence à analyser ne sera pas nécessairement retrouvée à travers toutes les lectures disponibles (notion de « couverture »). Si, sur un fragment de 25 Mb, seulement 20 Mb comportent une information de séquence fiable, on dit que la couverture est de 80 p. 100.
La suite de cet article est accessible aux abonnés
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Véronique BLANQUET : professeure de génétique, université de Limoges
- Nathalie DUPRAT : ingénieure d'études en techniques biologiques
- Lionel FORESTIER : ingénieur d'études en expérimentation et techniques biologiques
Classification
Médias

Exemple de diversité au sein d’un gène
Encyclopædia Universalis France

Détermination classique de la séquence d’un fragment d’ADN
Encyclopædia Universalis France
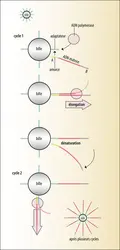
Principe de la PCR en émulsion (Ion Torrent®)
Encyclopædia Universalis France
Voir aussi
- AMORCEUR DE POLYMÉRISATION
- HÉRÉDITAIRES MALADIES ou MALADIES GÉNÉTIQUES
- NUCLÉOTIDES
- ADN POLYMÉRASE
- ARN MESSAGER ou ARNm
- CYTOSINE
- ADÉNINE
- RÉPLICATION, biologie moléculaire
- SCIENCES HISTOIRE DES, XXe XXIe s.
- SÉQUENÇAGE, génétique moléculaire
- GÉNOME
- ARN NON CODANTS ou ARNnc
- BIOLOGIE MOLÉCULAIRE
- INFORMATION GÉNÉTIQUE
- EXPRESSION GÉNÉTIQUE
- EXON
- GÉNÉTIQUE MOLÉCULAIRE
- MOLÉCULES BIOLOGIQUES, structure et fonction
- BIOLOGIE HISTOIRE DE LA
- MICROBIOTES
- MÉTAGÉNOMIQUE
