
- 1. L’ADN, molécule support de l’information génétique
- 2. Le séquençage « historique et classique » de l’ADN : la méthode de Sanger
- 3. Le séquençage NGS et ses approches à haut débit
- 4. La troisième génération de séquençage
- 5. Le single cell sequencing (SCS)
- 6. Ordonner les données de séquençage
- 7. Exploiter les données de NGS
- 8. Évolution des techniques et perspectives
- 9. Bibliographie
SÉQUENÇAGE HAUT DÉBIT DE L'ADN
Exploiter les données de NGS
Le séquençage à haut débit permet de réaliser plusieurs types d’analyses, selon bien sûr l’information biologique recherchée (variants, variation d’expression…), mais aussi la taille, la complexité du génome étudié et son information disponible dans les bases de données…
Séquençage de novoet reséquençage
Le séquençage de novo permet de reconstruire un génome n'ayant pas de référence répertoriée dans les bases de données. Les génomes de l’homme ou de nombreuses espèces très étudiées – animaux domestiques, certaines plantes ou bactéries – sont en effet déjà bien connus, mais de nombreux autres sont encore à étudier.
Le reséquençage de génome est réalisé lorsque le génome étudié possède déjà une référence mais que l’on veut approfondir ces connaissances (identification de nouveaux polymorphismes dans une race animale, par exemple). Le séquençage ciblé (autre approche de reséquençage) permet d’analyser spécifiquement des régions d'intérêt du génome. Il existe deux manières de sélectionner des zones précises à séquencer, soit par enrichissement (en capturant par hybridation complémentaire les régions d’intérêt à l’aide d’un mélange plus ou moins important d’oligonucléotide), soit par amplification ciblée. Ce ciblage peut être obtenu en utilisant une solution commerciale prête à l’emploi, c’est souvent le cas en recherche clinique, par exemple pour l’analyse des gènes BRCA1 et BRCA2 (gènes dont les mutations prédisposent au cancer du sein) à l’aide de systèmes génériques (panels d’amorces permettant d’amplifier et de séquencer simultanément toutes les régions comportant les mutations déjà identifiées, par exemple). Il peut aussi être réalisé à façon et est utilisé très fréquemment en recherche fondamentale. Ainsi, cette approche « custom » par enrichissement a été employée pour mener une étude d’association sur une région de 4,5 Mb associée au développement musculaire chez la race bovine limousine. La zone cible de l’ADN du génome bovin a été capturée chez les individus de l’étude à l’aide d’un panel de 55 000 sondes.
L’étude de l’exome
L’étude de l’exome (ensemble des exons du génome) permet d’analyser uniquement les zones codantes des gènes (les exons) et d’identifier, par exemple, des modifications communes de type petites insertions ou délétions – dites indels – et substitutions de base, qui entraînent des mutations non-sens ou faux-sens. En optimisant l’utilisation des capacités de séquençage disponibles, il est assez facile de percevoir l’intérêt budgétaire du séquençage des seules régions codantes qui, en soi, répond directement à beaucoup de problématiques liées uniquement à des modifications sur ces régions.
Le séquençage ChIP
De même, ces technologies permettent de cibler les zones du génome qui, à travers une interaction ADN/protéines, interviennent dans la régulation de l’expression des gènes. Une déclinaison particulière du séquençage NGS répond à cette problématique, c’est le séquençage ChIP, plus couramment dénommé « ChIP-Seq » pour chromatine immunoprecipitationsequencing. Il combine, d’une part, l’immunoprécipitation des zones de l’ADN sur lesquelles se trouvent fixées des protéines qui sont ciblées à l’aide d’anticorps spécifiques et, d’autre part, le séquençage haut débit de l’ADN afin d’identifier les régions d’intérêt ainsi sélectionnées (souvent promotrices de l’expression des gènes). Il peut être utilisé pour une cartographie précise de sites de liaison pour une protéine donnée.
Le séquençage de l’ARN
Le « DNA-Seq » permet de décoder la séquence ADN du génome, qui est normalement la même dans toutes les cellules d’un organisme, sauf éventuellement dans un contexte tumoral. Le séquençage NGS du transcriptome[...]
La suite de cet article est accessible aux abonnés
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Véronique BLANQUET : professeure de génétique, université de Limoges
- Nathalie DUPRAT : ingénieure d'études en techniques biologiques
- Lionel FORESTIER : ingénieur d'études en expérimentation et techniques biologiques
Classification
Médias

Exemple de diversité au sein d’un gène
Encyclopædia Universalis France
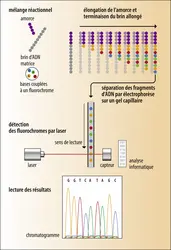
Détermination classique de la séquence d’un fragment d’ADN
Encyclopædia Universalis France
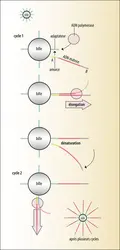
Principe de la PCR en émulsion (Ion Torrent®)
Encyclopædia Universalis France
